| Причина поломки | Устранение или предотвращение |
| Шум в коробке передач | |
| Износ подшипников, зубьев шестерен и синхронизаторов или их поломка | Заменить изношенные детали |
| Недостаточный уровень масла в коробке передач | Долить масло. Проверить и при необходимости устранить причины утечки масла Проверить и при необходимости устранить причины утечки масла |
| Некачественное масло в коробке передач | Заменить масло |
| Осевое перемещение валов | При необходимости заменить детали, фиксирующие подшипники или сами подшипники |
| Износ втулок оси шлицевого вала заднего хода | Заменить втулки шлицевого вала |
| Затруднённое переключение передач | |
| Неполное выключение сцепления | Проверка и ремонт сцепления |
| Заедание поверхности сферического шарнира | Снять рычаг и зачистить сопрягающиеся поверхности сферического шарнира |
| Деформация рычага переключения передач | Снять рычаг, устранить деформацию или заменить рычаг |
| Тугое движение штоков вилок (заусеницы, загрязнение, заклинивание блокировочных сухарей) | Разобрать, выявить причину, при необходимости отремонтировать или заменить изношенные детали |
| Неисправность синхронизаторов | Заменить изношенные детали или синхронизатор в сборе |
| Картер заправлен маслом несоответствующей марки | Слить масло, промыть коробку передач и заправить маслом или маслом-заменителем, рекомендуемым производителем |
| Выправить вилки, при необходимости заменить | |
| Ослабление затяжки или отвертывание винтов головок механизма переключения | Завернуть и закрепить винты |
| Разбиты отверстия под штифты в горловине механизма переключения | Заменить крышку механизма переключения или отремонтировать, расточив отверстия и запрессовав ступенчатые штифты |
| Самопроизвольное выключение или нечёткое включение передач | |
| Неправильное включение передач | При выжатой педали сцепления рычаг переключения перемещать до упора |
| Износ шариков или потеря упругости пружин фиксаторов штоков переключения передач | Снять крышку фиксаторов и осмотреть детали; при необходимости заменить |
| Износ или неправильное положение блокировочных сухарей штоков переключения передач | Разобрать и заменить изношенные детали, следя за правильностью сборки |
| Износ блокирующих колец синхронизаторов | Заменить изношенные кольца синхронизатора |
| Заменить пружины | |
| Износ зубьев муфты синхронизатора или зубчатого синхронизатора шестерни | Заменить муфту или шестерни |
| Ослабление затяжки гаек крепления коробки передач к картеру сцепления или гаек крепления удлинителя к картеру коробки передач | Затянуть гайки |
| Износ вкладышей управления переключением или износ резиновых деталей в рычагах переключения передач | Заменить изношенные детали |

 Оно теряет свои первоначальные свойства и перестает выполнять свои функции.
Оно теряет свои первоначальные свойства и перестает выполнять свои функции.Неисправности механической коробки передач и их устранение
Каждому автовладельцу известно стремление сохранить свой автомобиль в идеальном состоянии. Однако это практически невозможно, ведь необходимость его эксплуатации при любой погоде, а также при самых разных условиях влечет постоянный износ узлов и деталей машины, а также периодические поломки, которые требуется оперативно устранять.
Виды неисправностей механической трансмиссии автомобилей
Внутренне строение механизма механического переключения передач имеют свои особенности, при этом его неисправности могут делиться как на поломки самой коробки, так и на неисправности механизма трансмиссии.
Чтобы при возникновении любого вида неисправностей суметь восстановить работу автомобиля, следует вовремя обнаружить их и знать основные приемы по их устранению. Особенно это важно, если поломка настигла на трассе, в дороге, – тогда будет важно возобновить работу машины до момента визита к специалисту по ремонту.
Перечислим основные проявления неисправностей.
Как обнаружить неисправность?
К наиболее распространенным проявлениям неполадок в действии МКПП следует отнести следующие:
- Возникновение посторонних шумов при расположении рукоятки на нейтрали;
- Шумы во включенном состоянии, а также при начале работы;
- Затруднения при переключении скоростей;
- Выключение передачи происходит без воздействия на рычаг переключения;
- Наблюдается подтекание масла из трансмиссии.
МКПП: причины появления основных неисправностей и способы их устранения
Изучим более подробно основные виды неисправностей как самой коробки, так и механизма трансмиссии, а также оказание автомобилю «скорой помощи» в этих случаях.
Шум при положении рычага переключения в нейтральной позиции переключателя скоростей. Причина его появления чаще всего состоит в увеличении износа подшипников, расположенных в ведущем вале автомобиля, а также вследствие критически пониженного уровня масла в самой коробке передач. Также масло в трансмиссии может уже иметь слишком низкий уровень качества.
Устранить данное проявление можно проверкой уровня масла, последующей заменой изношенного подшипника. Если же трансмиссионное масло действительно давно не менялось, следует слить старое, затем заменить его на новое, которое будет соответствовать автомобилю. Старое масло рекомендуется проверить на наличие металлических посторонних частиц, воды, что является недопустимым.
Если же посторонние шумы слышны при переключении скоростей, то причиной этого может также одна из перечисленных выше причин либо деформация и повышенная степень износа блокирующего элемента, а также недостаточная устойчивость резьбовых соединений, неисправность синхронизаторов и не до конца выполненное выключение сцепления.
Устранение постороннего шума в момент включения передач:
- Следует в первую очередь проверить полную исправность сцепления;
- При неисправности синхронизаторов измерить величину зазора между шестернями и расположенным здесь же блокирующим кольцом, а также проверить шестерни на наличие повреждений;
Замер зазора между шестерней и кольцом
- При недостатке масла следует его долить;
- При не качественности трансмиссионного масла необходимо его полностью заменить на новое;
- Полностью выключить сцепление и повторить попытку переключения скоростей.
Когда шум слышен в процессе работы коробки, причиной этого может быть высокий износ шестерен, муфт синхронизаторов, подшипников, а также недостаточность уровня масла в трансмиссии.
Устранение шума может произойти при доливании масла, если уровень его ниже критического. Это можно обнаружить по наличию следов подтекания масла из трансмиссии, а профилактически следует заменять масло в автомобиле любой модификации каждые 10 000 км. Также необходимо проверить состояние муфт на предмет изношенности синхронизаторов и при необходимости их заменить.
Также необходимо проверить состояние муфт на предмет изношенности синхронизаторов и при необходимости их заменить.
Вот так выглядит изношенная шестерня с синхронизатором
Если же переключение передач осуществляется с затруднениями, здесь возможно ослабление крепления троса привода или его повреждение, не полностью выключенное сцепление, а также повреждение или сильный износ штока переключения скоростей.
Для лучшего переключения скоростей в МКПП следует в первую очередь проверить полноту отключения сцепления, проверить уровень масла и его качество (при непригодности для дальнейшего использования заменить на новое), а также проверить, не заедают ли шестерни и произвести тщательный осмотр на предмет наличия повреждений в системе рычагов переключения передач.
Не забываем проверять уровень масла
Рычаг переключения передач с трудом меняет положение, приходится прилагать определенные усилия для переключения скоростей. Причина здесь в первую очередь в недостаточном уровне масла в трансмиссии, ведь именно оно во многом отвечает за плавность всех движений, связанных с трансмиссией.
Для устранения этой неисправности следует проверить уровень масла в трансмиссии, а в случае необходимости его долить до установленного уровня.
В случае, когда передачи самопроизвольно переключаются, возможно, произошло уменьшение прочности резьбового соединения в креплении самой коробки, заел трос привода, либо излишне увеличился износ шестерен, достигла предела изношенность муфт синхронизаторов, а также шлицевых соединений, штока или вилки переключения передач.
Селектор передач с тросовым приводом
Для устранения этой неисправности необходимо почистить от загрязнений расстояние между корпусом двигателя и расположенным здесь же рычагом сцепления, внимательно осмотреть резьбовые соединения трансмиссии, при необходимости их подтянуть, а также при излишнем износе синхронизаторов либо шлицевых соединений произвести их полную замену.
Когда в механизме коробки переключения начинает подтекать масло, это видно даже невооруженным глазом. Причиной подтекания может стать недостаточность соединения самой трансмиссии либо изношенность сальников. Сальники при необходимости требуется заменить, резьбовые соединения же проверить и подтянуть.
Сальники при необходимости требуется заменить, резьбовые соединения же проверить и подтянуть.
Течь масла КПП
Перечисленные выше рекомендации по устранению неисправностей как самой коробки передач, так и механизма трансмиссии не исключают, а скорее обязывают владельца показать свой автомобиль специалисту и провести необходимые ремонтные работы в условиях автомастерской.
Неисправности любой из систем или отдельных узлов могут возникнуть в любой машине. Однако для предотвращения их появления следует соблюдать простые, но достаточно эффективные правила, касающиеся эксплуатации автомобиля. Их простота не означает неэффективность, ведь и здесь применимо правило, что любое заболевание легче и быстрее предотвратить, чем потом лечить.
По поводу автомобиля можно сказать, что легче, а также проще произвести профилактический осмотр, чем потом – дорогой ремонт.
Итак, приведем основные советы по предотвращению появления основных неисправностей автомобиля.
Рекомендации по сохранению здоровья механической трансмиссии
Как для начинающих автовладельцев, так и для имеющих солидный стаж езды, перечисленные ниже правила помогут как можно дольше избежать серьезных поломок механической коробки и быть уверенным к надежности своего автомобиля.
- Бережное отношение к коробке. Именно этот пункт является основополагающим, так как от метода его эксплуатации во многом зависит сохранность трансмиссии. Плавное переключение скоростей, без рывков и напряжения, — всё это сделает работу коробки более равномерной и движение автомобиля более спокойным.
- Периодическая проверка отсутствия потеков трансмиссионного масла и соблюдать его необходимый уровень. Проще всего это делать при планомерной замене масла в трансмиссии. Это правило также важно, поскольку незаметная на первый взгляд утечка масла может стать причиной серьезных поломок и огромных финансовых затрат.
- Если всё же ремонт коробки передач стал необходим, рекомендуется для него применять детали, которые имеют гарантию качества и подходят именно к марке вашего автомобиля.
 От того, из каких комплектующих состоит такой важный элемент, как коробка передач, зависит как качество ее работы, так и длительность эксплуатации.
От того, из каких комплектующих состоит такой важный элемент, как коробка передач, зависит как качество ее работы, так и длительность эксплуатации.
 От того, из каких комплектующих состоит такой важный элемент, как коробка передач, зависит как качество ее работы, так и длительность эксплуатации.
От того, из каких комплектующих состоит такой важный элемент, как коробка передач, зависит как качество ее работы, так и длительность эксплуатации.Заботясь о состоянии своего автомобиля, вы всегда будете с удовольствием совершать поездки, не беспокоясь о неожиданных неприятностях и будучи уверенными в своей безопасности и безопасности своих близких.
Хороших вам дорог!
Шум в коробке передач(если выжать сцепление, то он уменьшается/пропадает) |
|
| Причина | Метод решения |
| Недостаточно масла в картере КП | Проверить уровень масла и долить его при необходимости. Проверить утечку масла. |
| Масло в КП плохого качества, либо в него попала вода | Попадание воды в масло КП можно определить по наличию эмульсии беловатого цвета на щупе. Заменить масло в КП (см. какое лучше выбрать). Установить брызговик двигателя. Надеть трубку на сапун КП и вывести ее наверх, в защищенное от брызг место. какое лучше выбрать). Установить брызговик двигателя. Надеть трубку на сапун КП и вывести ее наверх, в защищенное от брызг место. |
| Подшипники, зубья или шестерни износились, либо повреждены | Заменить подшипники и шестерни. |
Не включается передача коробки передач(либо включается с трудом и посторонние шумы отсутствуют) |
|
| Причина | Метод решения |
| Деформирована тяга привода механизма переключения передач, либо реактивная тяга | Выправить или заменить тяги. |
| Ослабли винты крепления шарнира, хомута или рычага штока выбора передач | Затянуть винты, используя анаэробный герметик для резьб. |
| Сломались пластмассовые детали механизма переключения | Заменить детали. |
| Неправильная регулировка привода | Отрегулировать привод.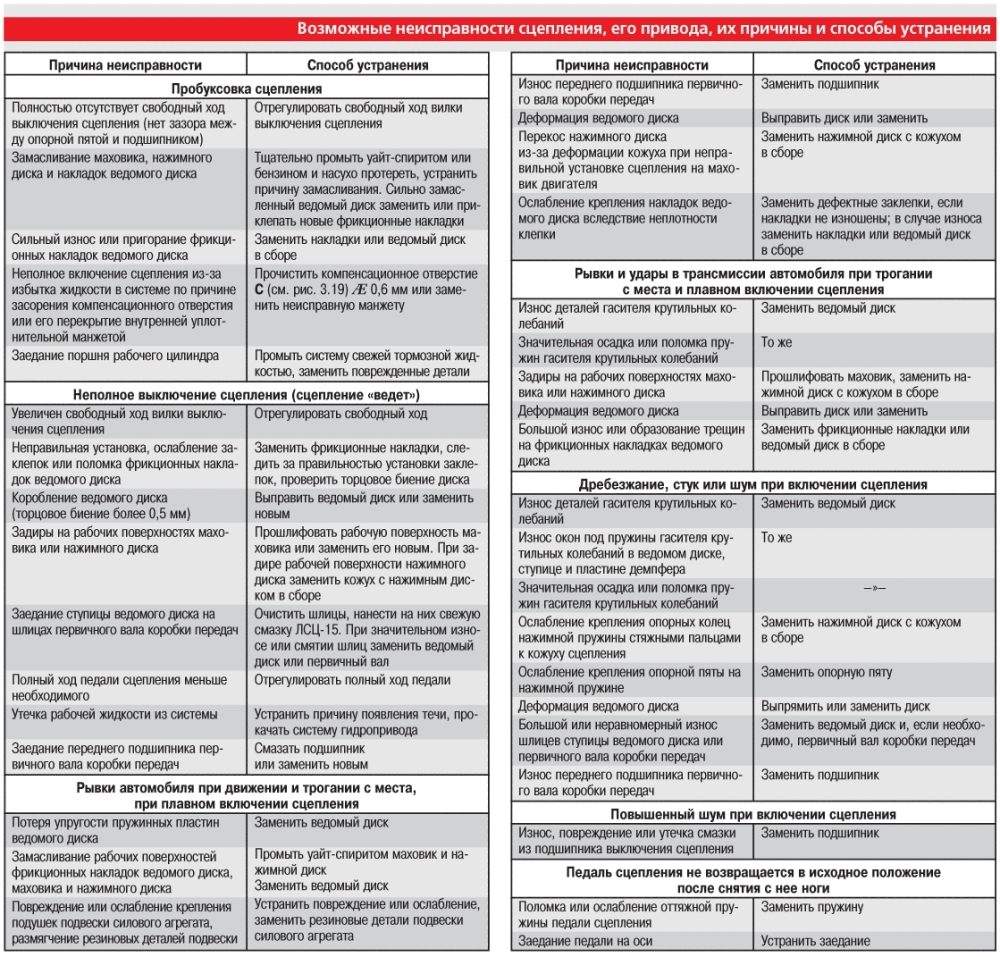 |
| Сломаны пружины механизма выбора передач, деформированы его детали | Заменить пружины, выправить детали или заменить механизм в сборе. |
| Ослабление посадок вилок переключения передач на штоке | Подтянуть фиксаторы вилок на штоках. |
| Не затянуты гайки валов коробки передач | Затянуть гайки. |
| Не полностью выключается сцепление | См. признаки неисправностей сцепления. |
Передачи самопроизвольно выключаются |
|
| Причина | Метод решения |
| Повреждение или износ шлицев на муфте, шестерне или ступице синхронизатора | Заменить неисправные детали. |
| Неправильная регулировка привода | Отрегулировать привода. |
| Ослабли пружины в механизме выбора передач, изношены штоки | Заменить изношенные детали. |
| Не затянуты гайки валов коробки передач | Затянуть гайки. |
| Потеряли упругость или разрушились опоры силового агрегата (подушки ДВС) | Заменить подушки двигателя. |
При включении передачи шумит коробка передач(треск, визг шестерен) |
|
|
|
|
| Причина | Метод решения |
| Сцепление выключается не полностью | См. признаки неисправностей сцепления. |
| Нет масла в картере коробки передач | Проверить уровень масла и долить его при необходимости. Проверить утечку масла. Продуть сапун. |
| Повреждены подшипники, зубья шестерен | Заменить подшипники и шестерни. |
| Износ кольца синхронизатора включаемой передачи | Заменить кольцо. |
Шум в КПП при движении автомобиля |
|
| Причина | Метод решения |
| Износ или разрушение подшипников | Заменить подшипники. Отрегулировать предварительный натяг подшипников коробки дифференциала. Отрегулировать предварительный натяг подшипников коробки дифференциала. |
| Увеличен зазор в зацеплении шестерен главной передачи, изношены их зубья | Заменить шестерни. |
Утечка масла коробки передач |
|
| Причина | Метод решения |
| Износ сальников: первичного вала, ШРУСов, штока выбора передач, износ уплотнителя вала привода спидометра | Заменить сальники. Продуть сапун коробки передач. Статья. |
| Сильный износ, забоины на поверхностях валов в местах сопряжения с поверхностями сальников | Небольшие повреждения зачистить мелкой шкуркой и заполировать. Устанавливая новый сальник, можно немного недопрессовать его, не допуская перекоса (при необходимости подложив под него дистанционные прокладки толщиной до 1 мм), чтобы кромка манжеты работала по неизношенной части вала. При значительных повреждениях – заменить валы и манжеты. |
| Большой люфт первичного вала коробки передач | Проверить состояние подшипников вала, их посадочных поверхностей, затяжку гайки. Заменить изношенные детали. |
| Ослабло крепление картера сцепления и крышки коробки передач, поврежден слой герметика между их сопрягающимися поверхностями | Подтянуть резьбовые соединения. Очистить поверхности от старого герметика. Перед нанесением нового – обезжирить поверхность. |
| Неплотно завернуты сливная пробка, датчик заднего хода | Подтянуть сливную пробку, датчик. |
Шум и вибрации. Признаки неисправности коробки передач | Авто
Коробка переключения передач (КПП) — одна из ключевых деталей автомобиля. Поломка трансмиссии может привести к большим тратам на ремонт.
Зачастую многие путают неисправность механизмов сцепления, актуальную для механической коробки и роботизированной, с неисправностью КПП. При проблемах со сцеплением появляется неприятный запах, рывки автомобиля, гул, обороты двигателя растут, но скорость не увеличивается.
Как продлить срок службы трансмиссии и вовремя понять, что коробка переключения скоростей имеет неисправности – в материале «АиФ-Челябинск».
Скрежет и вибрации
С точки зрения физики коробка передач в автомобиле — это рычажная пошаговая система, которая передаёт энергию от двигателя колёсам. На сегодняшний день существует ряд разновидностей КПП – механическая, гидромеханическая (автоматическая), роботизированная, вариаторная. Каждый тип коробки работает по-разному, тем не менее, есть общие признаки неисправности трансмиссии.
«Стоит забеспокоиться и предпринять решительные меры как можно скорее, если вы слышите странные звуки, переключая скорость, или обнаружили подтекание масла из-под коробки передач. Также у машины могут появиться повышенные вибрации. Нормально функционирующая трансмиссия не подаёт ни один из этих признаков», — рассказывает руководитель холдинга эксклюзивного представителя автомасел в России Вячеслав Кондратьев.
Чтобы выяснить причину неисправности, специалисты советуют в первую очередь проверить уровень и цвет масла в трансмиссии. Низкий уровень и грязное масло в коробке могут повредить её.
Низкий уровень и грязное масло в коробке могут повредить её.
Специалисты говорят, что для правильной работы трансмиссии необходимо доливать масло строго по уровню. Если его будет мало, то вероятен перегрев – самый худший враг коробки передач.
Признаки низкого уровня жидкости в коробке передач:
- Пробуксовка при переключении передач (для АКПП)
- Ручка переключения передач включается тяжело
- Колебания при переключении ручки коробки
Кроме того, такие же признаки могут быть при попадании другой жидкости в коробку передач, например, антифриза.
Неподходящее масло
Трансмиссия может испортиться, если подобрано неподходящее масло.
«К современному маслу для коробки передач предъявляются следующие требования: хорошая текучесть в диапазоне температур применения, надёжная защита от износа, обеспечение отличной синхронизации, высокая устойчивость к окислению и сдвигу, низкая склонность к пенообразованию, хорошая совместимость с уплотнениями, высокая экономичность», — говорит Вячеслав Михайлович.![]()
Разные типы КПП нуждаются в специальных трансмиссионных маслах, рекомендованных заводом-производителем. При неправильном подборе масла начинаются проскальзывания и вибрации, увеличивается расход топлива, износ, масло может подгорать.
Изношенные подшипники
Если в коробке передач при включении скорости появляется скрежет, но при переключении в нейтральное положение звук исчезает, то это может говорить об износе подшипников. Если вовремя не заменить эти подшипники, то коробка может заклинить, что приведёт к дорогостоящему ремонту.
«Трансмиссию рекомендовано проверять в соответствие с требованием автопроизводителя. Она нуждается в систематическом визуальном осмотре. Нужно тщательно следить, чтобы масло не подтекало. Кроме того, туда не должна попадать грязь, иначе передачи будут переключаться хуже», — советует инженер-механик Андрей Семёнов.
Советы по продлению срока службы трансмиссии от эксперта:
- Проверять уровень трансмиссионного масла следует в соответствии с рекомендацией автопроизводителя. В сервисной книжке автомобиля указан тип жидкости и необходимое количество для замены
- Не рекомендуется буксировать другие транспортные средства автомобилем, который оборудован АКПП
- Рекомендуется ставить автомобиль на ручник во время парковки, чтобы перераспределить вес автомобиля на тормоза с трансмиссии
- После движения назад всегда нужно делать полную остановку, прежде чем поехать вперед
- Фильтр в трансмиссии необходимо менять каждый раз в момент замены жидкости АКПП
 В сервисной книжке автомобиля указан тип жидкости и необходимое количество для замены
В сервисной книжке автомобиля указан тип жидкости и необходимое количество для заменыСмотрите также:
все о ремонте коробок передач на СТО
НовостиРемонт коробки передач АКПП и МКПППроблемы в работе коробки передач ни в коем случае нельзя игнорировать, так как функционирование этого агрегата жизненно важно для безопасности движения. Практически каждый автовладелец хорошо знает свой автомобиль и может сразу заподозрить наличие неполадки по следующим признакам:
- При переключении передач рычаг двигается туго, приходится прикладывать больше усилий, чем обычно;
- Во время езды коробка издает странные звуки, постукивания, слышится гул;
- Одну или несколько передач «выбивает»;
- Сцепление начинает срабатывать вверху педали;
- Снижается тяга, появляются пробуксовки;
- Начинает подтекать масло.

Распространные причины поломки КПП
- Езда на повышенных передачах и малых оборотах;
- Использование масла низкого качества;
- Слишком редкое обслуживание КПП, особенно в старых автомобилях;
- Ошибки при выполнении ремонта и сервиса КПП, использование некачественных запчастей;
- Самостоятельный ремонт КПП и неправильная сборка;
- Попытки самостоятельно снять или поставить КПП без соблюдения технологии.
Как и любой другой механизм автомобиля, коробка переключения передач требует регулярной диагностики и грамотного обслуживания, ведь чем позже вы заметите поломку, тем дороже обойдется ремонт КПП в Минске. Если же вы упустили момент, когда случилась поломка, необходимо обратиться н станцию техобслуживания – только специалисты смогут быстро и точно определить источник неисправности и устранить его.
Ремонт автоматической коробки передач
Автоматическая коробка – одна из самых прочных и надежных, особенно в автомобилях последних моделей. Однако даже она может выйти из строя, если не соблюдать условия эксплуатации, предписанные производителем, и пренебрегать своевременным техническим обслуживанием. Чаще всего поломка АКПП выражается следующими «симптомами»:
Однако даже она может выйти из строя, если не соблюдать условия эксплуатации, предписанные производителем, и пренебрегать своевременным техническим обслуживанием. Чаще всего поломка АКПП выражается следующими «симптомами»:
- Шум, жесткие толчки при переключении передач;
- Сильная вибрация;
- Задержки при включении;
- И прочие общие для всех коробок передач признаки неисправности.
Основными причинами поломки коробки-автомата являются агрессивное вождение, резкое торможение, низкий уровень масла и перегрев коробки, частое буксование и «цепная» поломка основных агрегатов из-за более мелкой неполадки. От сложности поломки и количества необходимых запчастей будет зависеть то, в какую сумму обойдется ремонт АКПП.
В перечень ремонтных работ могут входить:
- Замена масла;
- Снятие и установка;
- Разборка, дефектология, регулировка, наладка;
- Восстановление, замена, чистка гидроблока;
- Замена соленоидов;
- Замена мехатроника;
- Регулировка и замена исполнительных механизмов;
- Ремонт и замена гидротрансформатора;
- Ремонт ЭБУ;
- Устранение течи масла;
- Полная переборка, подгонка или замена запчастей.

В основном на СТО в Минске ремонт происходит следующим образом:
- Полная диагностика коробки;
- Снятие и переборка;
- Замена изношенных элементов, подгонка запчастей, устранение люфтов и течей;
- Сборка и тестирование коробки;
- Установка и отладка;
- Выходная диагностика.
Ремонт механической коробки переключения передач
Механика – проверенная десятилетиями и довольно надежная система, которой, однако, нужен тщательный уход и опытный взгляд мастера, чтобы еще на ранних стадиях определить поломки и устранить их, чтобы они не «потянули» за собой остальные узлы.
Чаще всего причиной неисправности коробки являются:
- Повреждение или износ шестерни передачи;
- Выход из строя подшипника первичного вала;
- Поломка кулисы или муфты;
- Срыв резьбы соединений агрегатов в коробке;
- Износ или повреждение троса привода;
- Износ вилки переключения передач;
- Износ или поломка блокиратора.

Вне зависимости от того, с какой поломкой вы столкнулись, ремонт МКПП лучше доверить профессионалам. При обращении на СТО в Минске вам проведут полную диагностику коробки передач, снимут и переберут ее, чтобы выявить все поврежденные элементы, проведут их полное восстановление или замену, сделают регулировку и наладку коробки и поставят ее на место. Непосредственно перед тем, как вернуть вам автомобиль, мастера проведут финишную диагностику – вы можете быть уверены, что коробка будет исправна и не подведет вас в самый ответственный момент!
Неисправности коробки передач в Daewoo Lanos
Автомобили Daewoo в Украине пользуются большим спросом. Это обусловлено их невысокой стоимостью и простым обслуживанием. Найти запчасти на Daewoo не сложно. Главное, выявить неисправность. Одна из наиболее нагруженных систем авто – механическая коробка передач и трансмиссия. Причины неисправностей бывают разные: износ подшипника или синхронизатора, падение уровня масла и попадание в него мусора, поломка муфты и деформация блокирующего устройства. Реже может потребоваться ремкомплект кулисы Ланос.
Реже может потребоваться ремкомплект кулисы Ланос.
Признаки поломанной МКПП
Трансмиссия отвечает за передачу крутящего момента от двигателя до колес. То есть без нее невозможно представить движение автомобиля. Главная роль в этом процессе отведена коробке переключения передач – КПП. Если она выходит из строя, то машину невозможно привести в движение.
На Daewoo Lanos установлена механическая коробка переключения передач. Ее также называют ручной. О неисправности КПП свидетельствуют следующие признаки:
посторонние звуки при работе;
невозможность переключения передач – всех или одной;
трудности во включении передач;
отключение передачи без участия водителя;
утечка масла.
Эти симптомы могут свидетельствовать о поломке следующих деталей:
Кроме того, сбои в работе КПП могут происходить из-за потери герметичности сальников, отворачивания гаек и болтов. Их износ может произойти из-за длительной эксплуатации, использования низкокачественного масла, отсутствия технического обслуживания автомобиля и агрессивного стиля вождения.
Их износ может произойти из-за длительной эксплуатации, использования низкокачественного масла, отсутствия технического обслуживания автомобиля и агрессивного стиля вождения.
Замена износившегося узла выполняется при полной разборке КПП. Эти работы лучше выполнять на СТО. Там же проведут диагностику КПП для выявления сломанного элемента.
Как продлить срок службы КПП
Долговечность и стабильность эксплуатации трансмиссии зависят от условий эксплуатации КПП. Чтобы продлить срок службы этой части авто, необходимо плавно переключать передачи. При этом важно, чтобы был полный выжим сцепления. Если выключение сцепления будет неполным при переключении передач, то быстро износятся шестерни.
Стиль вождения также играет немаловажную роль. Крайне важно включать передачу в соответствии со скоростным режимом. По возможности старайтесь двигаться на прямой передаче. Чаще всего, она значится под номером четыре. При таком режиме используется минимум шестерен и всего два вала. Это создает минимальную нагрузку на детали КПП. Как следствие, уменьшается вероятность их износа.
Это создает минимальную нагрузку на детали КПП. Как следствие, уменьшается вероятность их износа.
Каждый водитель должен уделить должное внимание обслуживанию КПП. Для этого необходимо:
Осматривать КПП после длительных поездок. Это нужно, чтобы вовремя выявить потеки масла. Они могут появиться возле сливной и заливной пробки, крышки КПП, на месте стыка коробки и двигателя.
Проверять уровень масла. Проверку нужно проводить каждые 10-15 тыс. км пробега.
Неисправности коробки передач ВАЗ 2110
регулировка тросика сцепления на ВАЗ 2110, 2111, 2112.
декада: езда задним ходом поломка кпп.
Как проводится ремонт коробок передач.
Кулиса ваз 2110 и замена сальника.
Переделка коробки передач на жигулях.
ВАЗ 2110 снятие КПП.
Отводим коробку передач от двигателя и снимаем ее.
кулисы на ВАЗ 2101-07.
Ремонт кпп и акпп своими руками — cartore ru.
Третья, модернизированная КПП ВАЗ 2110, собрана вместе с дифференциалом.
коробки передач ВАЗ 2110 2111 2112.
Устранение дребезга рычага КПП на Ваз 2110.
Ремонт КПП ваз 2110.
на автомобилях ваз 2108 ваз 2109 ваз 21099.
неисправноси синхронизатора ваз.
Особенности конструкции коробки передач ВАЗ 2170 Лада Приора.
кпп ваз 2109: ремонт. тест-драйвы автомобилей.
тест-драйвы автомобилей.
Какие неисправности могут быть у КПП ВАЗ 2110.
Преимущества КПП 12JS160TA.
Фото коробки передач ВАЗ 2110, novosel. ru.
ru.
переключения передач, шаровой опоры 17, тяги 14, штока выбора передач 5 и м…
Неисправности КПП ВАЗ 2110, ВАЗ 2111, ВАЗ 2112.
Кулиса КПП ВАЗ 2110.![]()
3.2.3. Разборка коробки передач.
ВАЗ 2110 Снятие и установка отделки.
КОРОБКА ПЕРЕДАЧ.
1 — задняя крышка картера коробки передач 2 — ведущая шестерня V передачи 3. ..
..
Подшипники для коробки ваз.
Схема дифференциала ваз 2110.
неисправность! масло насоса нет давления ваз 21103.
Неисправности кожуха сцепления и картера кпп.
Ремонт коробки передач ВАЗ 2101 — ВАЗ 2121.
Особенности и неисправности.
ваз 2110 схема коробка передач Мир схем.
(см. устанавливаемого в гнезде картера коробки передач под наружным кольцом…
Схема сборки коробки передач уаз 469.
Как завтулить стартер ваз.
Ремонт коробки передач на авто.
ведущая главной передачи; 4 — роликовый подшипник вторичного вала; 5 — масл. ..
..
ВАЗ 2110 — ремонт своими руками, видео и руководства.
Видео кпп ваз 2104 в разборе.
Возможные неисправности коробки передач, их причины и методы устранения.
Схема переключения коробки передачь ваз 2110.
неисправности холодильника lg no frost.
Природа контрольных точек клеточного цикла: факты и заблуждения | Journal of Biology
Существование критических «триггеров» или «точек невозврата» в ключевых переходах клеточного цикла было постулировано Мазиа еще в 1961 г. [2], хотя, как он позже признал в 1987 г., концепция изначально формулировка оказалась непродуктивной. Проблема заключалась в том, что триггеры считались важными внутренними компонентами молекулярных каскадов, управляющих клеточным циклом. Контрольные точки, напротив, представляют собой внешние механизмы контроля, которые не требуются для продвижения вперед [3].Таким образом, фундаментальной особенностью контрольно-пропускного пункта является то, что его деятельность не проявляется в условиях, в которых возможность ошибок минимальна: только когда условия становятся напряженными и вероятны ошибки, контрольно-пропускные пункты становятся важным инструментом выживания. Этот критерий лег в основу ранних скринингов для идентификации компонентов митотических контрольных точек у дрожжей [4]. Название трех хорошо известных белков митотических контрольных точек, Mad1-3, происходит от аббревиатуры «Mitotic Arrest Deficient», что отражает тот факт, что мутанты Mad проходят митоз с одинаковой кинетикой независимо от того, присутствует веретено или нет (и, следовательно, в наличие неприкрепленных кинетохор, которые в норме останавливают митоз — см.
[2], хотя, как он позже признал в 1987 г., концепция изначально формулировка оказалась непродуктивной. Проблема заключалась в том, что триггеры считались важными внутренними компонентами молекулярных каскадов, управляющих клеточным циклом. Контрольные точки, напротив, представляют собой внешние механизмы контроля, которые не требуются для продвижения вперед [3].Таким образом, фундаментальной особенностью контрольно-пропускного пункта является то, что его деятельность не проявляется в условиях, в которых возможность ошибок минимальна: только когда условия становятся напряженными и вероятны ошибки, контрольно-пропускные пункты становятся важным инструментом выживания. Этот критерий лег в основу ранних скринингов для идентификации компонентов митотических контрольных точек у дрожжей [4]. Название трех хорошо известных белков митотических контрольных точек, Mad1-3, происходит от аббревиатуры «Mitotic Arrest Deficient», что отражает тот факт, что мутанты Mad проходят митоз с одинаковой кинетикой независимо от того, присутствует веретено или нет (и, следовательно, в наличие неприкрепленных кинетохор, которые в норме останавливают митоз — см. легенду к рис. 1).Напротив, клетки дикого типа останавливаются в митозе, когда образование веретена ингибируется ядами микротрубочек. Однако в нормальных условиях клетки как дикого типа, так и Mad-дефицитные или организмы с низким числом хромосом и эффективными механизмами сборки веретена (например, дрожжи и дрозофилы ) растут одинаково хорошо, что отражает тот факт, что митотическая контрольная точка не важно, когда частота ошибок, естественно, низка.
легенду к рис. 1).Напротив, клетки дикого типа останавливаются в митозе, когда образование веретена ингибируется ядами микротрубочек. Однако в нормальных условиях клетки как дикого типа, так и Mad-дефицитные или организмы с низким числом хромосом и эффективными механизмами сборки веретена (например, дрожжи и дрозофилы ) растут одинаково хорошо, что отражает тот факт, что митотическая контрольная точка не важно, когда частота ошибок, естественно, низка.
Некоторые утверждают, что функция митотической контрольной точки у дрожжей и Drosophila отличается от таковой у млекопитающих, поскольку у млекопитающих инактивация генов контрольных точек приводит к летальному исходу даже при отсутствии других стрессов.Этот аргумент концептуально ошибочен, поскольку наблюдаемая разница в судьбах просто отражает различия в скорости сборки шпинделя. Из-за стохастической природы взаимодействия кинетохор и микротрубочек веретена присутствие многочисленных хромосом и/или центросом (полюсов веретена) значительно увеличивает время, необходимое для сборки веретена. При этом условии, если митотический выход не задерживается контрольной точкой до тех пор, пока все кинетохоры не прикрепятся к веретену, потомство будет анеуплоидным.По этой причине инактивация митотической контрольной точки у млекопитающих приводит к быстрому росту анеуплоидии и, в конечном итоге, к смерти [5]. Хорошей иллюстрацией взаимодействия между checkpoint, кинетикой сборки веретена и жизнеспособностью клетки/организма является недавняя работа с Drosophila , которые накапливают сверхштатные центросомы. Хотя само по себе это состояние не ставит под угрозу жизнеспособность, оно замедляет скорость сборки веретена. Как и ожидалось, устранение митотической контрольной точки путем делеции Mad2, которая не действует на Drosophila дикого типа, становится летальной для мух с нештатными центросомами [6].Важно подчеркнуть, что в последнем случае клетки не «убиты контрольной точкой», как это иногда описывается в литературе. Вместо этого клетки умирают, потому что в конечном итоге они становятся сильно анеуплоидными при отсутствии функциональной контрольной точки.
При этом условии, если митотический выход не задерживается контрольной точкой до тех пор, пока все кинетохоры не прикрепятся к веретену, потомство будет анеуплоидным.По этой причине инактивация митотической контрольной точки у млекопитающих приводит к быстрому росту анеуплоидии и, в конечном итоге, к смерти [5]. Хорошей иллюстрацией взаимодействия между checkpoint, кинетикой сборки веретена и жизнеспособностью клетки/организма является недавняя работа с Drosophila , которые накапливают сверхштатные центросомы. Хотя само по себе это состояние не ставит под угрозу жизнеспособность, оно замедляет скорость сборки веретена. Как и ожидалось, устранение митотической контрольной точки путем делеции Mad2, которая не действует на Drosophila дикого типа, становится летальной для мух с нештатными центросомами [6].Важно подчеркнуть, что в последнем случае клетки не «убиты контрольной точкой», как это иногда описывается в литературе. Вместо этого клетки умирают, потому что в конечном итоге они становятся сильно анеуплоидными при отсутствии функциональной контрольной точки. Функция митотической контрольной точки состоит в предотвращении преждевременного выхода из митоза и ни в чем другом.
Функция митотической контрольной точки состоит в предотвращении преждевременного выхода из митоза и ни в чем другом.
Неспособность отличить истинные белки контрольных точек от тех, которые участвуют в пути, на который нацелена митотическая контрольная точка, является обычным явлением и обычно является результатом слишком узкого внимания к молекулярным взаимодействиям без учета концептуального контекста.Очевидно, что белки контрольных точек должны взаимодействовать не только с отслеживаемой структурой или событием (например, с неприкрепленной кинетохорой), но также с путями и структурами, активность которых необходима для запуска прогрессии клеточного цикла. В этом случае, поскольку сама контрольная точка не требуется для продвижения вперед, белки, мутации которых продлевают митоз, никогда не могут считаться истинными компонентами контрольной точки.
Например, для выхода из митоза требуется активация крупной убиквитинлигазы, называемой комплексом, способствующим анафазе (APC) или циклосомой, который помечает для разрушения белки, которые удерживают реплицированные хромосомы вместе или удерживают клетку в митозе (к ним относятся секурин и циклин В; рисунок 1). APC активируется белком-активатором Cdc20, и если Cdc20 истощается, клетка останавливается в митозе. Несмотря на то, что Cdc20 напрямую взаимодействует с добросовестными белками контрольной точки (например, Mad2), этот фенотип ясно демонстрирует, что сам Cdc20 не является белком контрольной точки. Ожидается взаимодействие между Cdc20 и белками контрольных точек, поскольку целью митотической контрольной точки является биохимическое предотвращение преждевременной активации APC путем секвестрации Cdc20. Ошибочное рассмотрение Cdc20 или других белков, внутренне необходимых для прямой митотической прогрессии, как непосредственно вовлеченных в контрольную точку, приводит к деградации концепции контрольной точки обратно к внутренним «триггерам» Mazia.
APC активируется белком-активатором Cdc20, и если Cdc20 истощается, клетка останавливается в митозе. Несмотря на то, что Cdc20 напрямую взаимодействует с добросовестными белками контрольной точки (например, Mad2), этот фенотип ясно демонстрирует, что сам Cdc20 не является белком контрольной точки. Ожидается взаимодействие между Cdc20 и белками контрольных точек, поскольку целью митотической контрольной точки является биохимическое предотвращение преждевременной активации APC путем секвестрации Cdc20. Ошибочное рассмотрение Cdc20 или других белков, внутренне необходимых для прямой митотической прогрессии, как непосредственно вовлеченных в контрольную точку, приводит к деградации концепции контрольной точки обратно к внутренним «триггерам» Mazia.
Репликация ДНК, Контрольная точка, Синтез ДНК
Авраам, RT Передача сигналов контрольной точки клеточного цикла через киназы ATM и ATR. Genes Dev 15 , 2177–2196 (2001).
Агилера, А. и Гомес-Гонсалес, Б. Геном
нестабильность: механистический взгляд на ее причины и последствия. Nat Rev Genet 9 , 204–217 (2008).
Геном
нестабильность: механистический взгляд на ее причины и последствия. Nat Rev Genet 9 , 204–217 (2008).
Annunziato, A. T. Раздельное решение: что происходит с нуклеосомы во время репликации ДНК? Дж Биол Хим 280 , 12065–12068 (2005).
Бодди, М. Н. и Рассел, П. Репликация ДНК пропускной пункт. Курс. биол. 11 , R953–R956 (2001).
Branzei, D. & Foiani, M. Взаимодействие репликации контрольные точки и восстанавливающие белки в остановившихся вилках репликации. Восстановление ДНК (Amst) 6 , 994–1003 (2007).
Branzei, D. & Foiani, M. Регуляция репарации ДНК на протяжении всего клеточного цикла. Нат Рев Мол Cell Biol 9 , 297–308 (2008).
Branzei, D. & Foiani, M. Поддержание генома стабильность на вилке репликации. Нат Rev Mol Cell Biol 11 , 208–219 (2010).
Карр, А. М. Контрольные точки, зависящие от структуры ДНК, как
регуляторы репарации ДНК. Восстановление ДНК
(Амст) 1 , 983–994 (2002).
Восстановление ДНК
(Амст) 1 , 983–994 (2002).
Дурочер, Д. и Джексон, С. П. DNA-PK, ATM и ATR как датчики повреждения ДНК: вариации на тему? Curr Opin Cell Biol 13 , 225–231 (2001).
Грот, А. и др. Проблемы с хроматином во время репликации и восстановления ДНК. Cell 128 , 721–733 (2007).
Hartwell, L.H. & Weinert, TA. Контрольно-пропускные пункты: элементы управления, обеспечивающие порядок событий клеточного цикла. Наука 246 , 629–634 (1989).
Хеллер, Р. К. и Марианс, К. Дж. Сборка реплисом и прямой перезапуск застопорившихся вилок репликации. Nat Rev Mol Cell Biol 7 , 932–943 (2006).
Хеникофф, С., Фуруяма, Т. и Ахмад, К. Хистон варианты, сборка нуклеосом и эпигенетическое наследование. Trends Genet 20 , 320–326 (2004).
Кастан, М. Б. и Бартек, Дж. Контрольные точки клеточного цикла
и рак. Природа 432 , 316–323 (2004).![]()
Katou, Y. и др. Белки контрольной точки S-фазы Tof1 и Mrc1 образуют стабильную репликационную паузу. сложный. Природа 424 , 1078–1083 (2003).
Ламберт С., Фрогет Б. и Карр А. М. Арестованы обработка вилки репликации: взаимодействие между контрольными точками и рекомбинация. Восстановление ДНК (Amst) 6 , 1042–1061 (2007).
Леман, А. Р. и др. др. Human Timeless и Tipin стабилизируют репликационные вилки и облегчают слипчивость сестринских хроматид. J Клеточная наука 123 , 660–670 (2010).
Макфарлейн, Р. Дж., Миан, С. и Далгаард, Дж. З. многие аспекты роли семейств белков Тима-Типина в биологии хромосом. Cell Cycle 9 , 700–705 (2010).
Nasmyth, K. & Haering, CH Cohesin: его роли и механизмы. Annu Rev Genet 43 , 525–558 (2009).
Ногучи, Э. и др.
др. Swi1 и Swi3 являются компонентами комплекса защиты вилки репликации
в делящихся дрожжах. Mol Cell Biol 24 , 8342–8355 (2004).
Нюберг, К. А. и др. др. К ПОДДЕРЖАНИЮ ГЕНОМА: контрольные точки повреждения ДНК и репликации. Annu Rev Genet 36 , 617–656 (2002).
Полсен, Р. Д. и Симприч, К. А. Путь ATR: тонкая настройка вилки. Ремонт ДНК (Амст) 6 , 953–966 (2007).
Uhlmann, F. Вопрос выбора: установление сцепления сестринских хроматид. Отчеты EMBO 10 , 1095-1102 (2009 г.).
19.5: Нарушение контрольных точек клеточного цикла может вызвать рак
Если контрольная точка выходит из строя или если клетка подвергается физическому повреждению хромосом во время клеточного деления, или если она подвергается изнурительной соматической мутации в предыдущей фазе S , она может самоуничтожиться в ответ на последующую биохимическую аномалию.Это еще один пример апоптоза . С другой стороны, когда клетки умирают от внешнего повреждения, они подвергаются некрозу , случайной , а не запрограммированной смерти. В клетках, показанных ниже, химически индуцировали апоптоз или некроз, после чего идентифицировали как апоптоз или некроз с использованием флуоресцентных маркеров (йодид пропидия, зеленый; акридиновый оранжевый, оранжевый).
В клетках, показанных ниже, химически индуцировали апоптоз или некроз, после чего идентифицировали как апоптоз или некроз с использованием флуоресцентных маркеров (йодид пропидия, зеленый; акридиновый оранжевый, оранжевый).
Только зелено-флуоресцирующие (апоптотические) клетки в конечном итоге образуют апоптотические тельца.Напротив, некротические (флуоресцирующие оранжевым цветом) клетки теряют свои плазматические мембраны, не образуют таких «тел» и в конечном итоге распадаются. (400-кратное увеличение). Различия в ультраструктуре между некрозом и апоптозом также видны на электронных микрофотографиях колбочек и палочек (слева и справа соответственно) ниже. Звездочка указывает на цитоплазматическое набухание, характерное для некротического конуса. Белые стрелки указывают на ядра, характерные для апоптоза палочек.
Как мы уже отмечали, циклирующие клетки продолжают делиться до тех пор, пока не достигнут G 0 в терминально дифференцированном состоянии. Большинство терминально дифференцированных клеток очищаются апоптозом , когда они достигают конца своей эффективной жизни, чтобы быть замененными стволовыми клетками. Мы также отметили, что случайная передача сигналов может вывести клетки из G 0 , что приведет к возобновлению клеточной пролиферации. Хотя эти клетки явно аномальны, они не обнаруживаются механизмами защиты от апоптоза. Таким образом, они подвергаются неконтролируемому клеточному делению, становясь раковыми клетками. Точно так же физически поврежденные или мутировавшие клетки могут иногда не подвергаться апоптозному клиренсу.Когда они это сделают, они также могут стать раковыми клетками. Апоптотический клиренс и неконтролируемая пролиферация раковых клеток сравниваются ниже
Большинство терминально дифференцированных клеток очищаются апоптозом , когда они достигают конца своей эффективной жизни, чтобы быть замененными стволовыми клетками. Мы также отметили, что случайная передача сигналов может вывести клетки из G 0 , что приведет к возобновлению клеточной пролиферации. Хотя эти клетки явно аномальны, они не обнаруживаются механизмами защиты от апоптоза. Таким образом, они подвергаются неконтролируемому клеточному делению, становясь раковыми клетками. Точно так же физически поврежденные или мутировавшие клетки могут иногда не подвергаться апоптозному клиренсу.Когда они это сделают, они также могут стать раковыми клетками. Апоптотический клиренс и неконтролируемая пролиферация раковых клеток сравниваются ниже
346 Апоптоз (запрограммированная гибель клеток) и некроз
A. Белок P53 опосредует нормальный контроль клеточного цикла
Раковый рост может возникнуть, если нормальная делящаяся клетка подвергнется соматической мутации, которая нарушит нормальный контроль клеточного цикла. Подумайте, например, о чрезмерном выражении cdk . В качестве альтернативы представьте себе 90 177 уровней циклина 90 178 в дочерних клетках, которые никогда не падают; такие клетки никогда не перестанут циркулировать.
Подумайте, например, о чрезмерном выражении cdk . В качестве альтернативы представьте себе 90 177 уровней циклина 90 178 в дочерних клетках, которые никогда не падают; такие клетки никогда не перестанут циркулировать.
Другие возможности включают клетку в G 0 , которая стимулируется к повторному циклированию из-за неподходящей встречи с гормоном или другим сигналом. Если их не обнаружить, эти аномалии могут трансформировать клетки в раковые. Белок p53 (показан ниже) представляет собой ДНК-связывающий белок, регулирующий гены, который выявляет некоторые из этих аномалий и позволяет делящимся клеткам восстанавливать повреждения, прежде чем пройти через контрольные точки клеточного цикла… или, если это не удастся, приведет к апоптозу. клетки.
Неудивительно, что мутации в гене белка P53 (называемого у людей TP53 ) связаны со многими видами рака человека (поджелудочной железы, легких, почечных клеток, молочной железы и т. д.). Половина случаев рака у человека связана с мутацией p53 генов. Таким образом, p53 является одним из класса белков-супрессоров опухолей . Исследования людей с состоянием, известным как LFS ( синдром Ли-Фраумени ), выявили по крайней мере один мутантный аллель p53 .Мутация приводит к ~ 100% риску развития рака в течение всей жизни, начиная с детства. В культивируемых клетках мутагенизированные гены p53 проявляют ключевые характеристики раковых клеток, включая нерегулируемую клеточную пролиферацию и подавление апоптоза.
д.). Половина случаев рака у человека связана с мутацией p53 генов. Таким образом, p53 является одним из класса белков-супрессоров опухолей . Исследования людей с состоянием, известным как LFS ( синдром Ли-Фраумени ), выявили по крайней мере один мутантный аллель p53 .Мутация приводит к ~ 100% риску развития рака в течение всей жизни, начиная с детства. В культивируемых клетках мутагенизированные гены p53 проявляют ключевые характеристики раковых клеток, включая нерегулируемую клеточную пролиферацию и подавление апоптоза.
1. Как работает p53
Белок p53 в норме связан с активным белком Mdm2 . Чтобы включить контрольные точки клеточного цикла, p53-Mdm2 должен быть отделен и храниться отдельно, чтобы дать p53 время для действия. В делящихся клетках физический или химический стресс, такой как повреждение ДНК во время роста клеток, может активировать киназу ATM . Киназа ATM, в свою очередь, фосфорилирует Mdm2 , заставляя его диссоциировать от p53. Эта же киназа также фосфорилирует другую киназу, Chk2 , а также теперь «свободную» p53 . События, инициированные киназой ATM, более подробно описаны ниже.
Киназа ATM, в свою очередь, фосфорилирует Mdm2 , заставляя его диссоциировать от p53. Эта же киназа также фосфорилирует другую киназу, Chk2 , а также теперь «свободную» p53 . События, инициированные киназой ATM, более подробно описаны ниже.
Каждый из белков и ферментов, фосфорилируемых киназой ATM, играет роль в функционировании контрольной точки клеточного цикла и остановке клеточного цикла во время исправления ошибок:
- Теперь отделен от Mdm2, Phospho-p53 активно активирует несколько генов, включая ген p21 .
- Белок P21 связывается с cdks ; циклины не могут связываться с P21-cdks .
- Active Phospho-Chk2 катализирует фосфорилирование циклина; фосфоциклины не могут связываться с p21-cdks .
- Неспособность циклинов связывать cdks специфически блокирует клеточный цикл между фазами G 1 и S и G 2 6 7 90,75 M 90,75 M 90,75 M 90,75 M 90,75 M 90,75 M.
Эти опосредованные киназой события в контрольных точках клеточного цикла показаны ниже.
Клеточный цикл остается остановленным, пока клетка пытается завершить основные биохимические процессы, необходимые для исправления вызванных стрессом или других физических или химических аберраций, прежде чем перейти к следующей фазе цикла. Если репарация ДНК или другие исправления успешны, клетка может перейти к следующей фазе.
Если нет, протеасомы нацелены на деградацию комплекса Chk2-циклин .Аналогичным образом любой P53 , остающийся связанным с нефосфорилированным Mdm2 , также подвергается разрушению протеасомами. В результате любая клетка, неспособная исправить последствия стресса или химического повреждения или восстановить повреждение ДНК, становится мишенью для апоптоза .
Уровни и активность p53 , а также других белков, обсуждавшихся выше, контролируют как количество белка p53 , доступного для ответа на аномалии клеточного цикла, так и сами ответы. Фосфорилирование (активация) р53 приводит не только к быстрой остановке клеточного цикла, но и к активации генов, кодирующих белки, необходимые для репарации ДНК и белков, необходимых для апоптоза (в случае неудачи попыток репарации) . Взаимодействия p53 с различными белками, приводящие к альтернативным клеточным судьбам, суммированы ниже.
Фосфорилирование (активация) р53 приводит не только к быстрой остановке клеточного цикла, но и к активации генов, кодирующих белки, необходимые для репарации ДНК и белков, необходимых для апоптоза (в случае неудачи попыток репарации) . Взаимодействия p53 с различными белками, приводящие к альтернативным клеточным судьбам, суммированы ниже.
Таким образом, p53 подавляет рост злокачественных опухолей либо на
- позволяет репарировать ДНК или другие клетки до возобновления нормального клеточного цикла, предотвращая нерегулируемые клеточные деления; после восстановления p53 и другие белки инактивируются и/или разрушаются, и клеточный цикл может возобновиться.
- Неспособность восстанавливать/исправлять проблемы клеточного цикла запускает события, ведущие к апоптозу, тем самым также блокируя онкогенез путем уничтожения поврежденных клеток.
Теперь должно быть ясно, почему мутант p53 , который уменьшает или устраняет выработку белка p21 или блокирует выработку необходимого белка репарации ДНК, позволяет поврежденным клеткам проникать в S и поддерживать их репликацию и деление, превращая их в раковые клетки. Интересно, что по сравнению с людьми от рака умирают лишь немногие киты или слоны, несмотря на то, что у них в тысячи раз больше клеток, чем у людей. Причина, по-видимому, в том, что, по крайней мере, у слонов целых 20 копий (40 аллелей) генов p53! Таким образом, мутация в одном аллеле одного из них может оказывать незначительное влияние, в то время как преобладают опухоле-репрессирующие эффекты остальных генов р53. Прочтите об этом недавнем исследовании на сайте Киты и слоны не болеют раком!
Интересно, что по сравнению с людьми от рака умирают лишь немногие киты или слоны, несмотря на то, что у них в тысячи раз больше клеток, чем у людей. Причина, по-видимому, в том, что, по крайней мере, у слонов целых 20 копий (40 аллелей) генов p53! Таким образом, мутация в одном аллеле одного из них может оказывать незначительное влияние, в то время как преобладают опухоле-репрессирующие эффекты остальных генов р53. Прочтите об этом недавнем исследовании на сайте Киты и слоны не болеют раком!
2.Центральное место действия p53 в регуляции клеточного цикла
Из-за его многочисленных ролей в регулировании и содействии репарации ДНК, а также в контроле контрольных точек клеточного цикла, p53 был назван « Хранителем Генома »! Вот еще одно свидетельство этой центральной роли.
а) «Онкогенные вирусы»
Вирусы, вызывающие рак, включают вирус папилломы человека ( HPV ), вирус Эпштейна-Барра ( EBV ), вирус иммунодефицита человека ( HIV ), вирусы гепатита B и C ( HBV, HCV ), вирус герпеса человека 8 ( HHV-8 ) и обезьяний вирус 40 ( SV40 ).
Доказана связь между SV40, p53 и раком. SV40 является вирусным контаминантом вакцин против полиомиелита, которые использовались в 1960-х годах. Вирус является онкогенным у млекопитающих, хотя связь SV40 и рака у людей не доказана. В инфицированных клетках ДНК SV40 проникает в ядро, где может интегрироваться в геном клетки-хозяина. Инфекции SV40 обычно латентны (т. е. не причиняют вреда). Однако активация может привести к клеточной трансформации и росту злокачественных сарком в мышцах, а также опухолей в других органах.РНК-полимераза II в инфицированных клетках транскрибирует гены SV40, продуцируя белки, которые реплицируют и инкапсулируют вирусную ДНК в мембрану для создания новых вирусных частиц. Однако относительно небольшой геном SV40 не кодирует все ферменты и факторы, необходимые для репликации вирусной ДНК. Инфицированные клетки сами обеспечивают эти факторы, продуцируя их только во время фазы S . В это время большой Т-антиген SV40 (выработанный вскоре после заражения) проникает в ядро клетки-хозяина, где он регулирует транскрипцию генов, необходимых для репликации вируса и образования вирусных частиц. Большой Т-антиген также связывается с р53 , препятствуя транскрипции белков, гены которых регулируются р53 . Неспособная выполнять функции контрольной точки, клетка-хозяин бесконтрольно делится, образуя раковые опухоли. Дерегуляция клеточного цикла большим Т-антигеном обеспечивает переход к S-фазе и нерегулируемую корепликацию ДНК вируса и клетки-хозяина.
Большой Т-антиген также связывается с р53 , препятствуя транскрипции белков, гены которых регулируются р53 . Неспособная выполнять функции контрольной точки, клетка-хозяин бесконтрольно делится, образуя раковые опухоли. Дерегуляция клеточного цикла большим Т-антигеном обеспечивает переход к S-фазе и нерегулируемую корепликацию ДНК вируса и клетки-хозяина.
б) р53 и передача сигнала
Стресс может активировать пути передачи сигнала.Например, мутации, влияющие на сигнальный путь MAPK (MAP kinase), могут приводить к онкогенезу. Это может быть объяснено наблюдением, что при активации путь MAPK приводит к усиленной продукции киназы, которая фосфорилирует p53 . Активный фосфо-р53 , в свою очередь, усиливает активацию пути передачи сигнала МАРК. Вы можете вспомнить, что передача сигнала MAPK обычно заканчивается митогенным ответом.
Другим примером взаимодействия p53 является белок FAK (киназа фокальной адгезии ). Активность FAK повышается за счет интегрина опосредованной передачи сигнала. Напомним, что мембранные интегрины связывают фибронектин , способствуя образованию внеклеточного матрикса, или ВКМ . Повышенная активность FAK участвует в регуляции межклеточной и межклеточной адгезии ВКМ в фокальных точках адгезии . Другая роль FAK заключается в непосредственном связывании с неактивным p53 и увеличении связывания p53-Mdm2. Как мы только что видели, устойчивый p53-Mdm2 нацелен на убиквитинирование… и окончательное уничтожение! Фактически, аномально высокие уровни FAK связаны со многими различными линиями опухолевых клеток (толстая кишка, молочная железа, щитовидная железа, яичники, меланома, саркома…).Это происходит, когда p53 не может должным образом активировать контрольные точки клеточного цикла.
Активность FAK повышается за счет интегрина опосредованной передачи сигнала. Напомним, что мембранные интегрины связывают фибронектин , способствуя образованию внеклеточного матрикса, или ВКМ . Повышенная активность FAK участвует в регуляции межклеточной и межклеточной адгезии ВКМ в фокальных точках адгезии . Другая роль FAK заключается в непосредственном связывании с неактивным p53 и увеличении связывания p53-Mdm2. Как мы только что видели, устойчивый p53-Mdm2 нацелен на убиквитинирование… и окончательное уничтожение! Фактически, аномально высокие уровни FAK связаны со многими различными линиями опухолевых клеток (толстая кишка, молочная железа, щитовидная железа, яичники, меланома, саркома…).Это происходит, когда p53 не может должным образом активировать контрольные точки клеточного цикла.
В то время как взаимодействия, подразумеваемые здесь, сложны и активно изучаются, эта активность p53 определенно подтверждает его центральную роль как защитника генома и защитника клеточного деления .
B. Рост и поведение раковых клеток
Различные типы раковых клеток имеют разные ростовые и другие поведенческие свойства. Возможно, вы слышали о медленно растущих и быстрорастущих видах рака. Рак толстой кишки обычно медленно растет. Периодические колоноскопии , выявляющие и удаляющие колоректальные опухоли у людей среднего и пожилого возраста, могут предотвратить заболевание (хотя риски заболевания и сама процедура должны быть сбалансированы). Рак поджелудочной железы быстро растет и обычно остается незамеченным, пока не достигнет поздней стадии. Двумя целями медицинских исследований являются обнаружение различных видов рака на достаточно ранней стадии для успешного вмешательства и, конечно же, поиск эффективных методов лечения.
Единственная мутировавшая клетка в ткани может стать точкой роста опухоли , по существу массы клеток, клонированных из исходной мутировавшей. Доброкачественные опухоли или новообразования (например, груди и матки миомы у женщин или обычные родинки у любого из нас) перестают расти и не опасны для жизни. Их часто удаляют хирургическим путем для удобства пациента (или потому, что клетки в некоторых других доброкачественных опухолях могут стать раковыми).
Доброкачественные опухоли или новообразования (например, груди и матки миомы у женщин или обычные родинки у любого из нас) перестают расти и не опасны для жизни. Их часто удаляют хирургическим путем для удобства пациента (или потому, что клетки в некоторых других доброкачественных опухолях могут стать раковыми).
Злокачественные опухоли (также называемые злокачественными новообразованиями ) являются злокачественными и могут расти за пределами самой опухоли. Когда опухолевые клетки выделяются, они могут попадать в кровоток и перемещаться в другие части тела, явление, называемое метастазированием . Раковые клетки, которые метастазируют, могут стать очагом образования новых опухолей во многих различных тканях. Поскольку раковые клетки продолжают циклически повторять свою ДНК, они могут подвергаться еще большему количеству соматических мутаций.Эти дальнейшие изменения могут способствовать метастазированию и росту раковых клеток в разных частях тела.
C. Стратегии лечения рака
Существует множество различных видов рака, возникающих в различных тканях организма. Все они разделяют свойство неконтролируемого клеточного деления, хотя и по разным молекулярным и не всегда понятным причинам. Две основные стратегии лечения рака, разработанные в 20 веке, направлены на то, чтобы каким-то образом нарушить репликацию.
- Лучевая терапия основана на том факте, что большинство клеток в нашем организме не делятся, направляя мутагенное излучение на опухоли в надежде, что реплицирующаяся ДНК будет мутирована во многих местах (т. е. в генах), что опухолевые клетки больше не смогут выжить или правильно воспроизвести.
- Химиотерапия используется для борьбы с опухолями, плохо реагирующими на облучение или труднодоступными для лучевых технологий, а также для борьбы с раковыми заболеваниями, которые даже не образуют очаговых опухолей (таких как лимфомы и лейкемии с вовлечением лимфоцитов и клеток крови) . Эти химиотерапевтические также направлены на нарушение репликации или митотической активности. Например, вспомните кордицепин (дидезоксиаденозинтрифосфат или ддАТФ). Присутствуя во время репликации, ddATP включается в растущую цепь ДНК, после чего к цепи ДНК не могут быть добавлены дополнительные нуклеотиды. Это делает ddATP мощным химиотерапевтическим разрушителем репликации. Taxol — еще один химиопрепарат, который действует в данном случае не за счет ингибирования репликации S-фазы, а за счет блокирования деполимеризации микротрубочек веретенообразных волокон, тем самым блокируя митотические анафазы и телофазы в последней части М- и С-фаз цикла. Колхицин (растительный алкалоид) атакует раковые (и другие делящиеся) клетки, блокируя образование микротрубочек и тем самым предотвращая образование веретенообразных волокон в митотической профазе.
 Эти химиотерапевтические также направлены на нарушение репликации или митотической активности. Например, вспомните кордицепин (дидезоксиаденозинтрифосфат или ддАТФ). Присутствуя во время репликации, ddATP включается в растущую цепь ДНК, после чего к цепи ДНК не могут быть добавлены дополнительные нуклеотиды. Это делает ddATP мощным химиотерапевтическим разрушителем репликации. Taxol — еще один химиопрепарат, который действует в данном случае не за счет ингибирования репликации S-фазы, а за счет блокирования деполимеризации микротрубочек веретенообразных волокон, тем самым блокируя митотические анафазы и телофазы в последней части М- и С-фаз цикла. Колхицин (растительный алкалоид) атакует раковые (и другие делящиеся) клетки, блокируя образование микротрубочек и тем самым предотвращая образование веретенообразных волокон в митотической профазе.
Эти химиотерапевтические также направлены на нарушение репликации или митотической активности. Например, вспомните кордицепин (дидезоксиаденозинтрифосфат или ддАТФ). Присутствуя во время репликации, ddATP включается в растущую цепь ДНК, после чего к цепи ДНК не могут быть добавлены дополнительные нуклеотиды. Это делает ddATP мощным химиотерапевтическим разрушителем репликации. Taxol — еще один химиопрепарат, который действует в данном случае не за счет ингибирования репликации S-фазы, а за счет блокирования деполимеризации микротрубочек веретенообразных волокон, тем самым блокируя митотические анафазы и телофазы в последней части М- и С-фаз цикла. Колхицин (растительный алкалоид) атакует раковые (и другие делящиеся) клетки, блокируя образование микротрубочек и тем самым предотвращая образование веретенообразных волокон в митотической профазе. Эти методы лечения не эффективны против всех видов рака, и, конечно же, они не нацелены на определенные виды раковых клеток.![]() Их успех основан просто на том факте, что раковые клетки быстро и постоянно размножаются, в то время как другие типы клеток этого не делают. Многие, если не все, побочные эффекты облучения и химиотерапии являются результатом повреждения нормальных делящихся клеток (например,г., клетки волосяных фолликулов, ответственные за выпадение волос у многих больных раком, истощение клеток крови, которые не могут быть заменены стволовыми клетками в костном мозге).
Их успех основан просто на том факте, что раковые клетки быстро и постоянно размножаются, в то время как другие типы клеток этого не делают. Многие, если не все, побочные эффекты облучения и химиотерапии являются результатом повреждения нормальных делящихся клеток (например,г., клетки волосяных фолликулов, ответственные за выпадение волос у многих больных раком, истощение клеток крови, которые не могут быть заменены стволовыми клетками в костном мозге).
В настоящее время многие исследования сосредоточены на мобилизации собственной иммунной системы организма для создания более конкретных и целенаправленных методов лечения рака. В захватывающей истории более 100 лет назад доктор Уильям Б. Коли ввел неизлечимому больному раком стрептококковые бактерии, которые затем избавились от опухоли после выздоровления от инфекции (подробности см. в The Earlyest Cancer Immunotherapy Испытания).Феномен «Dr. Токсины Коли изначально считались противоопухолевым эффектом бактерий. Но к 1948 году его широко связывали с иммунным ответом, активируемым инфекцией. В 1990-х годах ученые вновь обратились к иммунному ответу на рак, и на рубеже 21-го века исследования иммунотерапии рака набрали обороты (и более существенное финансирование исследований!).
В 1990-х годах ученые вновь обратились к иммунному ответу на рак, и на рубеже 21-го века исследования иммунотерапии рака набрали обороты (и более существенное финансирование исследований!).
Недавние эксперименты по иммунотерапии на животных и клинические испытания на людях являются многообещающими. Несколько иммунотерапевтических препаратов уже одобрены U.S. FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов). Стратегии иммунотерапии рака основаны на том факте, что ваше тело иногда распознает маркеры раковых клеток (например, молекулы клеточной поверхности) как чужеродные, тем самым создавая иммунную защиту против этих клеток. Но этот ответ иногда недостаточно силен, чтобы уничтожить новые, быстро делящиеся раковые клетки. Рак, по-видимому, возникает, когда иммунный ответ слаб. Существуют разные, иногда пересекающиеся подходы к иммунотерапии рака. Все они основаны на том факте, что раковые клетки каким-то образом мутировали и производят аберрантные белки, которые иммунная система может воспринимать как достаточно чужеродные, чтобы вызвать иммунный ответ, каким бы слабым он ни был.![]() Некоторые виды иммунотерапии стремятся усилить этот иммунный ответ. Другие стремятся изолировать или создать уникальные антигены раковых клеток, которые будут иммунизировать пациента при введении этих раковых антигенов. Некоторые виды иммунотерапии приведены в таблице на следующей странице. Как видно из таблицы, иммунотаргетирование раковых клеток уже доказало свою высокую эффективность. В некоторых случаях терапия является примером персонализированной медицины , в которой лечение индивидуально адаптировано для вас как пациента.Проблемы с иммунотерапией заключаются в том, что
Некоторые виды иммунотерапии стремятся усилить этот иммунный ответ. Другие стремятся изолировать или создать уникальные антигены раковых клеток, которые будут иммунизировать пациента при введении этих раковых антигенов. Некоторые виды иммунотерапии приведены в таблице на следующей странице. Как видно из таблицы, иммунотаргетирование раковых клеток уже доказало свою высокую эффективность. В некоторых случаях терапия является примером персонализированной медицины , в которой лечение индивидуально адаптировано для вас как пациента.Проблемы с иммунотерапией заключаются в том, что
- они требуют много времени и труда, а их производство дорого.
- , хотя они могут «вылечить» вас, они, скорее всего, не подействуют на кого-то другого.
- Подобно облучению и химиотерапии, иммунотерапия имеет свои неприятные, а иногда и серьезные побочные эффекты.
Более подробное обсуждение иммунотерапии рака можно найти на веб-сайте Cancer.gov по адресу Cancer Treatment Immunotherpay.
ПРИМЕЧАНИЕ. Термин ингибитор контрольной точки в контексте иммунотерапии отличается от термина контрольные точки , описывающего порталы для прохождения эукариотического клеточного цикла.
Дисфункция иммунных контрольных точек при васкулите крупных и средних сосудов
Аутоиммунное воспаление, поражающее стенки аорты и ее боковых ветвей, приводящее к образованию, разрыву и расслоению аневризмы, а для сосудов среднего калибра — к окклюзии просвета. Чтобы защитить хозяина от таких опасных для жизни осложнений, слои стенок жизненно важных артерий иммунопривилегированы, что делает их устойчивыми к локализованным иммунным и воспалительным реакциям (53). Роль иммунопривилегии в стенке аорты нарушена при гигантоклеточном артериите (ГКА), аутоиммунном и аутовоспалительном заболевании, вызывающем аортит и артериит второй-пятой ветвей аорты (3, 44, 52).Клинические последствия включают повреждение стенки аорты, синдром дуги аорты и ишемические осложнения в глазах и задних отделах головного мозга. Почти всегда воспаление сосудов у пациентов с ГКА сочетается с синдромом системного воспаления, проявляющимся лихорадкой, задержкой развития, ревматической полимиалгией и избыточной продукцией острофазовых реагентов (15). ГКА диагностируют на основании биопсии височной артерии, типичных данных компьютерной томографии или магнитно-резонансной ангиографии.Терапевтически индукционная терапия по-прежнему основывается на высоких дозах кортикостероидов (52). Недавно к терапевтическому арсеналу были добавлены новые иммунодепрессанты, но они, по-видимому, не вызывают стойкой ремиссии заболевания (3, 50). Хронический вялотекущий васкулит в настоящее время представляет собой наиболее серьезную клиническую проблему, осложненную отсутствием данных о соотношении риск/польза хронической иммуносупрессии и неполным пониманием иммунопатологии персистирующего васкулита.
Почти всегда воспаление сосудов у пациентов с ГКА сочетается с синдромом системного воспаления, проявляющимся лихорадкой, задержкой развития, ревматической полимиалгией и избыточной продукцией острофазовых реагентов (15). ГКА диагностируют на основании биопсии височной артерии, типичных данных компьютерной томографии или магнитно-резонансной ангиографии.Терапевтически индукционная терапия по-прежнему основывается на высоких дозах кортикостероидов (52). Недавно к терапевтическому арсеналу были добавлены новые иммунодепрессанты, но они, по-видимому, не вызывают стойкой ремиссии заболевания (3, 50). Хронический вялотекущий васкулит в настоящее время представляет собой наиболее серьезную клиническую проблему, осложненную отсутствием данных о соотношении риск/польза хронической иммуносупрессии и неполным пониманием иммунопатологии персистирующего васкулита.
Иммунное поражение Т-клеток и макрофагов ГКА лишает артерию иммунных привилегий воспаление (рис.1). Как правило, пораженные ГКА артерии заполнены гранулематозными инфильтратами, состоящими из Т-клеток CD4, высокоактивированных макрофагов, многоядерных гигантских клеток и дендритных клеток (ДК) (53).
 Клоны Т-клеток CD4 с идентичными рецепторами Т-клеток были выделены из независимых биоптатов тканей пациентов с ГКА, что подтверждает концепцию васкулитогенного антигена (13, 56). Функциональный анализ тканевых цитокинов, продуцируемых при васкулитных поражениях, подтвердил представление о мультифункциональных Т-клетках, попавших в очаги поражения.Т-клетки, резидентные в сосудистой стенке, продуцируют интерферон (IFN)-γ, IL-17, IL-9 и IL-21, участвующие в росте Т-клеток-хелперов (Th)1, Th27, Th9 и фолликулярных Th (T FH ). в процессе болезни (рис. 1) (5, 6, 46, 51). Примечательно, что продукция IL-17 в тканях очень чувствительна к кортикостероидной терапии, тогда как Т-клетки, продуцирующие IFN-γ, по-видимому, не подвержены влиянию высоких доз системных стероидов (6, 53), что подтверждает концепцию функциональной гетерогенности среди пораженных популяций Т-клеток. Отсутствие регуляторных Т-клеток CD4 и CD8 в сосудистой стенке согласуется с наблюдением, что иммунные поражения ГКА вызываются несколькими типами эффекторных Т-клеток.
Клоны Т-клеток CD4 с идентичными рецепторами Т-клеток были выделены из независимых биоптатов тканей пациентов с ГКА, что подтверждает концепцию васкулитогенного антигена (13, 56). Функциональный анализ тканевых цитокинов, продуцируемых при васкулитных поражениях, подтвердил представление о мультифункциональных Т-клетках, попавших в очаги поражения.Т-клетки, резидентные в сосудистой стенке, продуцируют интерферон (IFN)-γ, IL-17, IL-9 и IL-21, участвующие в росте Т-клеток-хелперов (Th)1, Th27, Th9 и фолликулярных Th (T FH ). в процессе болезни (рис. 1) (5, 6, 46, 51). Примечательно, что продукция IL-17 в тканях очень чувствительна к кортикостероидной терапии, тогда как Т-клетки, продуцирующие IFN-γ, по-видимому, не подвержены влиянию высоких доз системных стероидов (6, 53), что подтверждает концепцию функциональной гетерогенности среди пораженных популяций Т-клеток. Отсутствие регуляторных Т-клеток CD4 и CD8 в сосудистой стенке согласуется с наблюдением, что иммунные поражения ГКА вызываются несколькими типами эффекторных Т-клеток.
Рис. 1. Иммунное поражение при гигантоклеточном артериите (ГКА). A : здоровая артерия среднего размера с открытым просветом и тремя слоями стенки. Сосудистые дендритные клетки (ДК) располагаются на границе медии и адвентиции, где они могут функционировать для защиты иммунных привилегий сосудистой стенки. B : ГКА вызывается гранулематозным воспалением в слоях артериальной стенки. Основными клеточными игроками являются Т-клетки CD4, высокоактивированные макрофаги и ДК. На разных участках стенки преобладают разные воспалительные пути.В прямоугольниках указаны типы клеток и их продукты, связанные с заболеванием, которые образуют адвентициальный инфильтрат и медиальный инфильтрат. Адвентиция является важным местом презентации антигена и активации Т-клеток. Несколько типов эффекторных Т-клеток располагаются в адвентиции и медии. Эффекторные функции макрофагов играют решающую роль в повреждении тканей и ответе на заживление ран, сосредоточенном в среде. Артерия реагирует на воспаление программой дезадаптивной реструктуризации. Неоангиогенез в адвентиции открывает шлюзы для инфильтрации воспалительных клеток.Мобилизация и миграция миофибробластов создают быстро прогрессирующую гиперплазию интимы, приводящую к окклюзии просвета и ишемическим осложнениям ГКА. TGF-β, трансформирующий фактор роста-β; Т, Т-клетка.
Артерия реагирует на воспаление программой дезадаптивной реструктуризации. Неоангиогенез в адвентиции открывает шлюзы для инфильтрации воспалительных клеток.Мобилизация и миграция миофибробластов создают быстро прогрессирующую гиперплазию интимы, приводящую к окклюзии просвета и ишемическим осложнениям ГКА. TGF-β, трансформирующий фактор роста-β; Т, Т-клетка.
Многие клетки, находящиеся в гранулеме, являются макрофагами, часто называемыми гистиоцитами. Их функциональное назначение тесно связано с их расположением в стенке сосуда (рис. 1) (48), что подчеркивает тот факт, что различные микроокружения выполняют разные функции в патологическом процессе. Макрофаги в гиперплазированной интиме являются провоспалительными, продуцируя индуцируемый оксид азота в васкулитных поражениях (57).Медиальные макрофаги участвуют в ремоделировании сосудистой стенки и защите от окислительного стресса, продуцируя факторы роста (тромбоцитарный фактор роста, VEGF и FGF), металлопротеиназы и альдозоредуктазы (17, 18, 39, 40). Адвентициальные макрофаги совместно продуцируют трансформирующий фактор роста-β 1 , IL-6 и IL-1β (57). Имеющиеся данные свидетельствуют о том, что ингибирование васкулитных макрофагов, эффективно достигаемое при лечении пациентов с ГКА глюкокортикоидами, не восстанавливает иммунные привилегии сосудистой стенки (6).Исследования повторных биопсий у пациентов до и после терапии глюкокортикоидами показали, что васкулитные инфильтраты сохраняются у большинства пациентов даже после 9-12 месяцев терапии (27).
Адвентициальные макрофаги совместно продуцируют трансформирующий фактор роста-β 1 , IL-6 и IL-1β (57). Имеющиеся данные свидетельствуют о том, что ингибирование васкулитных макрофагов, эффективно достигаемое при лечении пациентов с ГКА глюкокортикоидами, не восстанавливает иммунные привилегии сосудистой стенки (6).Исследования повторных биопсий у пациентов до и после терапии глюкокортикоидами показали, что васкулитные инфильтраты сохраняются у большинства пациентов даже после 9-12 месяцев терапии (27).
Доказательства презентации антигена в гранулемах основаны на активном участии ДК. Считается, что сосудистые дендритные клетки как местная популяция сосудистой стенки (22, 26, 36) играют критическую роль в защите иммунных привилегий артерии. Оснащенные костимулирующими лигандами и хемокинами, они контролируют клеточный поток и усиливают ответы Т-клеток (14, 22, 26, 32, 36, 55).ДК также являются источником иммуноингибирующих лигандов, передающих отрицательные сигналы Т-клеткам, что критически определяет размер и продолжительность иммунных ответов (20, 59). Недавние данные демонстрируют, что иммунная контрольная точка белка запрограммированной гибели клеток-1 (PD-1), через которую лиганд запрограммированной гибели клеток-1 (PD-L1) + DCs обеспечивает стоп-сигнал для PD-1 + Т-клеток, дефектен в GCA (62), что подчеркивает регуляторную важность пораженных DCs.
Недавние данные демонстрируют, что иммунная контрольная точка белка запрограммированной гибели клеток-1 (PD-1), через которую лиганд запрограммированной гибели клеток-1 (PD-L1) + DCs обеспечивает стоп-сигнал для PD-1 + Т-клеток, дефектен в GCA (62), что подчеркивает регуляторную важность пораженных DCs.
Что такое иммунные контрольные точки?
Протективный и патогенный Т-клеточный иммунитет представляет собой многоэтапный процесс, основанный на клональной селекции антиген-специфических клеток.При встрече с антигеном Т-клетки активируются и массово размножаются, увеличивая размер клона. Некоторые клетки разовьются в Т-клетки памяти и войдут в места хранения во вторичных лимфоидных тканях. Другие будут перемещаться в периферические ткани для повторной встречи с антигеном, выполнения прямых эффекторных функций или оказания помощи (посредством высвобождения цитокинов и мембранных лигандов) множеству иммунных эффекторных клеток (45, 61). Каждый из этих этапов лежит в основе регуляции путем уравновешивания стимулирующих и ингибирующих сигналов, что в конечном итоге приводит к регулированию амплитуды, продолжительности и качества иммунного ответа (рис. 2) (12). Классические костимулирующие сигналы происходят от взаимодействий CD28-CD80 или CD28-CD86, которые абсолютно необходимы для управления метаболическими программами размножения Т-клеток (8). Входящие сигналы «вперед» сочетаются с сигналами «стоп», часто называемыми иммунными контрольными точками. Хорошо известные стоп-сигналы, сдерживающие активацию и клональную экспансию Т-клеток, происходят от антигена-4, ассоциированного с цитотоксическими Т-лимфоцитами (CTLA-4; CD152) и PD-1 (CD279). Оба рецептора иммунных контрольных точек играют решающую роль в предотвращении чрезмерного иммунитета, защите хозяина от побочного повреждения и предотвращении аутоиммунитета (30, 38, 63).Оба рецептора в настоящее время признаны мишенями для иммунотерапии рака (33, 35, 37), поскольку опухоли научились узурпировать противоопухолевый иммунитет, усиливая передачу сигналов иммунных контрольных точек. В то время как считается, что CTLA-4 нарушает ранние этапы активации Т-клеток, PD-1 обычно активируется на Т-клетках после активации, предоставляя возможность ингибировать более поздние этапы программы клональной экспансии. Запуск PD-1 его лигандами PD-L1 и PD-L2 активирует фосфатазу-2, содержащую домен гомологии Src 2 фосфатазы (1), тем самым ингибируя ключевые киназы, передающие каскад активации Т-клеток.PD-L1 и PD-L2 экспрессируются на антигенпрезентирующих клетках, опухолевых клетках и тканевых клетках, что позволяет им определять силу и продолжительность иммунных реакций (рис. 2) (7, 9, 64).
2) (12). Классические костимулирующие сигналы происходят от взаимодействий CD28-CD80 или CD28-CD86, которые абсолютно необходимы для управления метаболическими программами размножения Т-клеток (8). Входящие сигналы «вперед» сочетаются с сигналами «стоп», часто называемыми иммунными контрольными точками. Хорошо известные стоп-сигналы, сдерживающие активацию и клональную экспансию Т-клеток, происходят от антигена-4, ассоциированного с цитотоксическими Т-лимфоцитами (CTLA-4; CD152) и PD-1 (CD279). Оба рецептора иммунных контрольных точек играют решающую роль в предотвращении чрезмерного иммунитета, защите хозяина от побочного повреждения и предотвращении аутоиммунитета (30, 38, 63).Оба рецептора в настоящее время признаны мишенями для иммунотерапии рака (33, 35, 37), поскольку опухоли научились узурпировать противоопухолевый иммунитет, усиливая передачу сигналов иммунных контрольных точек. В то время как считается, что CTLA-4 нарушает ранние этапы активации Т-клеток, PD-1 обычно активируется на Т-клетках после активации, предоставляя возможность ингибировать более поздние этапы программы клональной экспансии. Запуск PD-1 его лигандами PD-L1 и PD-L2 активирует фосфатазу-2, содержащую домен гомологии Src 2 фосфатазы (1), тем самым ингибируя ключевые киназы, передающие каскад активации Т-клеток.PD-L1 и PD-L2 экспрессируются на антигенпрезентирующих клетках, опухолевых клетках и тканевых клетках, что позволяет им определять силу и продолжительность иммунных реакций (рис. 2) (7, 9, 64).
Рис. 2. Костимулирующие и коингибирующие сигналы в Т-клеточной регуляции. Т-клетки взаимодействуют с антигенпрезентирующими клетками, чтобы распознать родственный им антиген. Т-клеточный рецептор (TCR) связывается с комплексом человеческий лейкоцитарный антиген (HLA)-антиген, запуская каскад активации Т-клеток.Сопутствующие взаимодействия рецептор-лиганд обеспечивают либо положительные сигналы (костимуляция), либо передачу отрицательных сигналов (коингибирование), в конечном итоге регулируя качество, интенсивность и продолжительность ответа Т-клеток. Показана основная костимулирующая молекула CD28, которая связывается с CD80 и CD86 для усиления каскада активации Т-клеток. Также показаны два основных ингибирующих пути. Ингибирующие рецепторы цитотоксического антигена-4, ассоциированного с Т-лимфоцитами (CTLA-4) и белка запрограммированной гибели клеток-1 (PD-1), экспрессируемые на Т-клетках, активируются их лигандами на антиген-презентирующей клетке и обеспечивают «остановку иммунного ответа». сигнал Т-клетке.
Также показаны два основных ингибирующих пути. Ингибирующие рецепторы цитотоксического антигена-4, ассоциированного с Т-лимфоцитами (CTLA-4) и белка запрограммированной гибели клеток-1 (PD-1), экспрессируемые на Т-клетках, активируются их лигандами на антиген-презентирующей клетке и обеспечивают «остановку иммунного ответа». сигнал Т-клетке.
Иммунная контрольная точка PD-1 в GCA
PD-L1 экспрессируется на антигенпрезентирующих клетках, некоторых стромальных клетках и опухолевых клетках (7, 9, 64). Профилирование экспрессии генов продемонстрировало высокую экспрессию транскриптов PD-L1 в здоровых, невоспаленных артериях человека (62), где этот иммунозащитный лиганд может способствовать иммунной привилегии. Наоборот, пораженные ГКА артерии имели отчетливо низкие уровни транскриптов PD-L1, а встроенные в стенку ДК не имели выраженной экспрессии PD-L1.Низкая экспрессия белка PD-L1 сохранялась для DC, генерированных ex vivo у пациентов с ГКА, тогда как DC, генерированные у пациентов с ревматоидным артритом (RA), реагировали на сигналы активации надежной индукцией PD-L1. Функциональное тестирование показало, что полученные от пациента PD-L1 low DC обеспечивают сильные сигналы, активирующие Т-клетки, а блокада PD-L1, вмешательство, которое, как известно, усиливает стимуляцию Т-клеток, не способно усиливать дальнейшие ответы Т-клеток. По сути, дефицит PD-L1 в ДК GCA оставляет у пациента не встречающие противодействия сигналы, активирующие Т-клетки (рис.3).
Функциональное тестирование показало, что полученные от пациента PD-L1 low DC обеспечивают сильные сигналы, активирующие Т-клетки, а блокада PD-L1, вмешательство, которое, как известно, усиливает стимуляцию Т-клеток, не способно усиливать дальнейшие ответы Т-клеток. По сути, дефицит PD-L1 в ДК GCA оставляет у пациента не встречающие противодействия сигналы, активирующие Т-клетки (рис.3).
Рис. 3. Дефектный иммунный контрольный пункт PD-1 при гигантоклеточном артериите. ДК от пациентов с ГКА экспрессируют высокие уровни костимуляторных лигандов CD80 и CD86, но низкие уровни ингибирующего лиганда запрограммированной гибели клеток-1 (PD-L1). Как следствие, они не могут ингибировать взаимодействующие Т-клетки. Неингибированные Т-клетки пролиферируют и приобретают множественные эффекторные функции. При васкулитных поражениях ГКА большинство Т-клеток несут рецептор PD-1. Пораженные Т-клетки многофункциональны, продуцируют провоспалительные цитокины [e. g., интерферон (IFN)-γ, IL-17 и IL-21] и способствуют реструктуризации сосудистой стенки за счет усиления микрососудистого неоангиогенеза и ускорения гиперплазии интимы.
g., интерферон (IFN)-γ, IL-17 и IL-21] и способствуют реструктуризации сосудистой стенки за счет усиления микрососудистого неоангиогенеза и ускорения гиперплазии интимы.
Как слабость стоп-сигналов, индуцированных PD-L1, влияет на Т-клеточный ответ при ГКА? В воспаленных височных артериях пациентов с ГКА практически все Т-клетки, попавшие в гранулемы, являются PD-1-положительными. PD-1 считается маркером отсроченной активации Т-клеток, распознающих антиген. Что еще более важно, присутствие этой поверхностной молекулы было связано с двумя исходами: истощением Т-клеток и активностью Th в зародышевом центре, обеспечиваемой так называемыми клетками PD-1 + T FH (43, 58).Ни один из этих двух сценариев не применим к гранулематозным поражениям при ГКА. Т-клетки, занимающие стенку воспаленного сосуда, определенно не истощены. Они сильно активируются, и была описана локальная продукция нескольких мощных Т-клеточных цитокинов. IFN-γ, по-видимому, особенно важен, и тканевой IFN-γ связан с интенсивностью процесса ремоделирования стенки (18). Пораженные Т-клетки также не ведут себя как клетки T FH ; они колокализуются с макрофагами, но не с В-клетками.Гранулематозные инфильтраты явно негативны для В-клеток, и у пациентов с ГКА не было выявлено продукции аутоантител (28).
Пораженные Т-клетки также не ведут себя как клетки T FH ; они колокализуются с макрофагами, но не с В-клетками.Гранулематозные инфильтраты явно негативны для В-клеток, и у пациентов с ГКА не было выявлено продукции аутоантител (28).
Функциональная активность Т-клеток PD-1 + в воспаленной артериальной стенке была исследована в модельной системе GCA, созданной путем пересадки нормальных человеческих артерий мышам с ослабленным иммунитетом (34, 62). Затем таких химерных мышей восстанавливают мононуклеарными клетками периферической крови, и в течение 1–2 недель развивается васкулит. Обработка таких химер человек-артерия-мышь антителом против PD-1 приводила к существенному усугублению воспаления стенки сосуда и инфильтрации Т-клеток (фиг.4). Анализ экспрессии тканевых генов выявил заметное усиление врожденного и адаптивного иммунитета после устранения взаимодействий PD-1/PD-L1. Трансплантаты артерий от химер, обработанных анти-PD-1, были обогащены широким спектром транскриптов цитокинов: IL-1β, IL-6, TNF-α, IL-23 p19, IL-27 p28, IL-7 и IL- 15. Эти данные наиболее совместимы с зависимой от PD-1 передачей сигналов, регулирующей находящиеся в тканях Т-клетки и макрофаги. Нарушение стоп-сигнала, производного от PD-1, в конечном итоге усугубляет множество эффекторных функций и в целом увеличивает интенсивность васкулитогенного иммунного ответа.
Эти данные наиболее совместимы с зависимой от PD-1 передачей сигналов, регулирующей находящиеся в тканях Т-клетки и макрофаги. Нарушение стоп-сигнала, производного от PD-1, в конечном итоге усугубляет множество эффекторных функций и в целом увеличивает интенсивность васкулитогенного иммунного ответа.
Рис. 4. Ингибирование контрольной точки при лечении антителами против PD-1 усугубляет васкулит. Человеческие артерии прививали иммунодефицитным мышам с диабетом NSG-γ без ожирения. Для индуцирования васкулита в привитых сосудах химерных мышей восстанавливали мононуклеарными клетками периферической крови пациентов с ГКА. Через две недели артерии человека были эксплантированы, и интенсивность васкулита определялась иммуноокрашиванием на CD3 + Т-клеток человека в срезах тканей.Перед забором человеческих артерий химерным мышам вводили антитела против PD-1 (100 мкг) или контрольный IgG путем внутрибрюшинных инъекций через день. Анти-CD3-связывающие Т-клетки (коричневые) в ткани визуализировали с помощью козьих антикроличьих вторичных антител, конъюгированных с пероксидазой хрена. По сравнению с контролем IgG ( слева ) блокада PD-1 ( справа ) заметно увеличивала плотность сосудистого Т-клеточного инфильтрата. Оригинальное увеличение: ×600.
Вероятно, наиболее важным наблюдением, сделанным на модели химеры артерий человека и мыши (62), было влияние нарушения сигнала PD-1 на ремоделирование воспаленной артериальной стенки.Увеличение плотности Т-клеток PD-1 + в васкулитных очагах приводило к усугублению интрамурального неоангиогенеза и индукции маркеров активации эндотелиальных клеток. Как правило, пораженные ГКА артерии имеют густую сеть новообразованных микрососудов, которые, как считается, обеспечивают быстрое привлечение воспалительных клеток и снабжение растущего воспалительного очага кислородом и питательными веществами. Более высокая плотность Т-клеток PD-1 + в артериальной стенке также была связана с размером интимального слоя, удваивая толщину интимы.Гиперплазия интимы вызывает ишемические осложнения ГКА, такие как слепота и инсульт (52, 54). Эти данные помещают передачу сигналов PD-1 и интенсивность активации Т-клеток на вершину процесса ремоделирования стенки. Дальнейшие исследования должны идентифицировать клеточных партнеров Т-клеток PD-1 + , например, эндотелиальные клетки, гладкомышечные клетки сосудов и другие, которые в конечном итоге вызывают патологические события.
Привязка дефицита иммуноингибиторного лиганда PD-L1 к сосудистым ДК поднимает вопрос о том, как это влияет на презентацию эндогенных и экзогенных антигенов.Васкулитогенные антигены при ГКА остаются неизвестными, а эпизодические сообщения об инфекционных антигенах, экспрессируемых в пораженных васкулитом артериях, не были подтверждены независимыми исследованиями. Привлекательная концепция эндогенных антигенов, вызывающих сосудистые заболевания, возникла при изучении гипертензии (16, 23, 29). При гипертонии повышенный окислительный стресс в ДК связан с образованием высокореактивных γ-кетоальдегидов (21, 60). Эти высокореактивные модификации белков связаны с изменением структуры и функции белков, а также с образованием неоантигенов, к которым иммунная система не толерантна.Последствия включают сильную активацию DC и индукцию продукции IL-6, IL-1β и IL-23, которые, в свою очередь, позволяют взаимодействующим Т-клеткам секретировать IL-17, IFN-γ и TNF-α. Неизвестно, работают ли подобные механизмы в средних и крупных артериях, подверженных васкулиту. Очевидно, ослабление функции контрольной точки из-за потери PD-L1 может действовать как петля амплификации, поддерживая иммунное распознавание аутоантигенов, которые иначе игнорируются.
В настоящее время неизвестно, является ли недостаточность контрольной точки PD-1 селективным дефектом при ГКА или имеет значение и при других аутоиммунных заболеваниях.Поскольку дефектный контрольный пункт PD-1, по-видимому, высвобождает иммунитет в стенке сосуда, было бы столь же важно изучить его роль в других сердечно-сосудистых заболеваниях, таких как атеросклероз и гипертония. Имеющиеся данные о мышиных моделях атеросклероза подтверждают концепцию о том, что интактный путь PD-1/PD-L1 необходим для подавления Т-клеток, которые способствуют образованию атеросклеротических поражений и воспалению бляшек (2, 11). Риск развития аневризмы аорты у пациентов с диагнозом ГКА удваивается по сравнению с контрольной группой того же возраста (42), что, вероятно, является прямым проявлением васкулитного поражения аорты.Кроме того, у лиц, страдающих ГКА, чаще диагностируют цереброваскулярные и сердечно-сосудистые заболевания, особенно у пожилых мужчин из неблагополучных социально-экономических районов (41). Интересно, что недавнее исследование (47) определило более низкий индекс массы тела в качестве предиктора риска ГКА, предполагая различные сценарии риска для тех, кто предрасположен к васкулиту крупных сосудов, по сравнению с теми, у кого классический атеросклероз.
Выводы, открытые вопросы и клинические последствия
В физиологических условиях стенки крупных артерий защищены от иммунной атаки.У пациентов с васкулитом крупных сосудов, таким как ГКА, эта привилегия нарушается, и иммунные клетки (особенно Т-клетки и макрофаги) проникают в слои стенки, чтобы инициировать ремоделирование стенки и, в конечном итоге, окклюзию просвета. Недавние исследования выявили дефекты в иммуноингибирующей передаче сигналов как предшествующие патологии при ГКА. В частности, находящиеся в тканях и генерируемые ex vivo ДК от пациентов с ГКА имеют дефект в усилении иммуноингибиторного лиганда PD-L1, таким образом, не в состоянии уравновесить стимулирующие сигналы ингибирующими сигналами.Таким образом, Т-клетки у пациентов с ГКА высвобождаются и способны проникать в стенку сосуда, рекрутировать макрофаги и образовывать гранулематозные скопления. Молекулярные механизмы, лежащие в основе дефектной индукции PD-L1, еще предстоит выяснить, но тесная корреляция между воспалительными маркерами и уровнями экспрессии DC PD-L1 у отдельных пациентов подчеркивает непосредственное влияние, которое может иметь этот молекулярный дефект (62). Действительно, накопление Т-клеток PD-1 + в сосудистых поражениях, по-видимому, имело прямые последствия для качества и интенсивности васкулитной реакции.Чем больше Т-клеток PD-1 + попадало в стенку сосуда, тем больше продуцировалось Т-клеточных цитокинов, тем больше образовывалось микрососудов и тем толще становился интимальный слой (табл. 1). Эти данные подтверждают новую патогенетическую модель ГКА, приписывающую окончательный контроль над процессами ремоделирования стенок активности Т-клеток. Не менее важным является признание того, что антиген-неспецифическая иммунная регуляция определяет исход сосудистого воспаления.
| 9061 | Клинические последствия | 9062
|---|---|
| T 10626 | |
| THECT INTERNY TRATIC INLARIZE | |
| Интерферон-γ Продукция IL-17 и IL-21 | |
| Продукция IL-7 и IL-15 | |
| Активность макрофагов | Продукция IL-1β, IL-6, IL-23 и TNF-α | Интрамуральный неоангиогенез | Плотность просвета микрососудов |
| Гиперплазия интимы | Толщина интимального слоя также поднимает несколько вопросов, которые будут иметь важное значение для дальнейших исследований.Фенотип PD-L1 low ДК ГКА, по-видимому, специфичен для заболевания, не влияя на ДК, генерируемые у пациентов с РА. Оба болезненных состояния считаются аутоиммунными и воспалительными. Эти наблюдения показывают, что воспалительная среда, даже если она присутствует в течение многих лет, недостаточна для изменения экспрессии PD-L1. В настоящее время проводятся эпигенетические исследования для оценки эпигенетического ландшафта промотора PD-L1 при ГКА, РА и ишемической болезни сердца. Большая часть информации о регуляции экспрессии PD-L1 получена в результате исследований опухолевых клеток.До сих пор механизмы, управляющие экспрессией PD-L1, поняты лишь поверхностно и, по-видимому, строго специфичны для типа клеток (4, 19, 24, 25, 31, 49). Недавние данные, собранные в опухолях со сверхэкспрессией PD-L1, предполагают роль новых структурных вариантов в 3′-нетранслируемой области промотора PD-L1 для определения поверхностной экспрессии (19). И IFN-γ, и LPS используются в стандартных тест-системах для усиления активности лиганда, а ДК пациентов с ГКА практически не реагируют на IFN-γ (62). Эти данные свидетельствуют о том, что IFN-зависимые сигнальные пути могут быть в первую очередь ответственны за дефект. Интересным аспектом выявления аномалий иммунных контрольных точек в патогенезе васкулита крупных сосудов является молекулярная взаимосвязь противоопухолевого иммунитета и иммунитета против стенок сосудов. Т-клетки CD4 у пациентов с васкулитом имеют низкий порог активации и могут выживать в тканевых нишах, из которых они иначе исключены. В отсутствие передачи сигналов PD-L1 они могут выполнять широкий спектр эффекторных функций, включая связь с сосудистыми клетками, такими как эндотелиальные клетки и миофибробласты.У пациентов с опухолями цитотоксические Т-клетки не допускаются в микроокружение опухоли, но восстанавливают доступ после ингибирования контрольной точки PD-1 (рис. 5) (35, 37, 64). Иммуноопосредованные побочные эффекты терапии ингибиторами контрольных точек связаны с развитием васкулита (10). Врачи и пациенты должны быть предупреждены о возможном риске антиваскулярного иммунитета как части растущего спектра иммунозависимых нежелательных явлений при иммунотерапии рака. Рис. 5. Иммунная контрольная точка PD-1 при раке и взаимодействие GCA между PD-L1 и PD-1 подает отрицательные сигналы Т-клеткам, эффективно подавляя эффекторные функции Т-клеток (см. рис.2). Опухолевые клетки ускользают от иммунного распознавания, экспрессируя иммуноингибиторный лиганд PD-L1. Т-клетки, пытающиеся убить раковые клетки, парализуются, получая негативные сигналы через свой рецептор PD-1. Недавние прорывы в иммунотерапии рака основаны на нацеливании на взаимодействие PD-L1/PD-1. Ингибиторы контрольных точек предотвращают взаимодействие PD-L1/PD-1 и привели к значительному успеху в повышении иммунитета к опухолям. Высвобождение Т-клеточного иммунитета у пациентов, получавших контрольные точки, связано с иммунологической токсичностью, т.е.г., индукция аутоиммунного заболевания. Сообщалось о лекарственно-индуцированном васкулите. При ГКА передача сигналов контрольных точек нарушена из-за низкой экспрессии PD-L1 на ДК стенки сосуда. Утрата этого «иммунного разрыва» позволяет Т-клеткам PD-1 + проникать в стенку сосуда и координировать воспалительную реакцию, повреждающую стенку. Т-клетки PD-1 + участвуют в регуляции выработки цитокинов в очагах поражения, управлении микрососудистым неоангиогенезом и усилении гиперплазии интимы, закупоривающей просвет.Учитывая дефицит отрицательной иммунной сигнализации, связанной с уклонением от ракового иммунитета, у пациентов с ГКА может быть очень эффективный противоопухолевый иммунный ответ. Множественные центросомы возникают из-за нарушения контрольной точки тетраплоидии и кластеров митотических центросом в клетках с скомпрометированным белком p53 и RBAbstractВысокая степень анеуплоидии характерна для большинства опухолей человека. Анеуплоидный статус может возникать из-за митотической недостаточности или недостаточности расщепления в сочетании с недостаточностью тетраплоидного контроля контрольных точек G 1 или из-за нарушения регуляции числа центросом, что приводит к изменению числа полюсов митотического веретена.р53 и белки кармана RB важны для контроля прогрессии G 1 , а ранее предполагалось, что р53 важен для контроля удвоения центросом. Мы демонстрируем здесь, что ни супрессия p53, ни семейства белков кармана RB не вызывает непосредственно изменения количества центросом ни в одной из нескольких первичных клеточных линий млекопитающих. Вместо этого увеличение числа центросом происходит в два этапа. Первым шагом является неспособность арестовать тетраплоидный контрольный пункт G 1 после неудачной сегрегации генома в митозе, а вторым шагом является кластеризация центросом на одном полюсе веретена в последующем тетраплоидном или анеуплоидном митозе.Триггером для этих событий является сбой митоза или расщепления, который не зависит от статуса p53 или RB. Наконец, мы находим, что фибробласты мышиных эмбрионов спонтанно входят в тетраплоид G 1 , что объясняет предыдущую демонстрацию центросомной амплификации за счет отмены р53 в этих клетках. Анеуплоидия и хромосомная нестабильность (CIN) характерны для подавляющего большинства опухолей человека (1) и связаны с прогрессирующим развитием инвазивных опухолей высокой степени злокачественности (2–4).Кроме того, высокая степень анеуплоидии коррелирует с плохим прогнозом (5–7). Недавние данные свидетельствуют о том, что анеуплоидия может быть необходимым промежуточным звеном в формировании многих солидных опухолей человека (8). Анеуплоидия может возникать по двум основным механизмам. Клетки могут пройти через тетраплоидный промежуточный процесс к мультиполярному митозу, который создает случайное распределение хромосом, или могут перейти непосредственно к анеуплоидии из-за отказа критического контроля эуплоидии. Хотя анеуплоидия может возникнуть непосредственно, тетраплоидия, по-видимому, является частым промежуточным этапом на пути к анеуплоидии.Во многих карциномах человека клетки с тетраплоидным содержанием ДНК возникают как ранний этап онкогенеза, предшествующий образованию анеуплоидных клеток (9, 10). Опухоли человека, такие как аденокарцинома пищевода (3, 11), карцинома шейки матки (12) и модельные системы опухолей грызунов (13, 14), проходят через тетраплоидные промежуточные соединения. Тетраплоидия может возникать либо из-за нарушения сегрегации хромосом во время митоза (15–18), либо из-за нарушения цитокинеза (19). Затем прогрессирование до анеуплоидии происходит в отсутствие p53-зависимого контроля контрольной точки, который обычно действует в G 1 для ареста тетраплоидных клеток. Альтернативно, анеуплоидия может возникать по механизмам, независимым от тетраплоидного промежуточного звена. Например, потеря контроля над дупликацией центросом, ведущая к аномальной амплификации центросом, будет создавать мультиполярные веретена и анеуплоидию. Дублирование центросом обычно происходит во время S фазы (20, 21) посредством cdk2-зависимого механизма (22-25) и находится под системой ограничений, которая гарантирует наличие одного и только одного события дупликации во время интерфазы. В результате точности дупликации нетрансформированная клетка имеет две центросомы при митозе, которые диктуют образование двух полюсов веретена.Если в интерфазе происходит более одного события дупликации, может получиться мультиполярное веретено, и геном будет сегрегировать анеуплоидным образом. Сообщалось, что p53 (26) и aurora A (27) участвуют в регуляции удвоения центросом во время интерфазы, поскольку аномалии числа центросом могут быть результатом нарушения функции любого белка. Однако ни p53, ни aurora A не оказывают убедительного влияния на удвоение центросом в течение одного клеточного цикла.Действительно, Meraldi et al. (28) недавно представили доказательства того, что ни aurora A, ни p53 напрямую не контролируют количество центросом во время S-фазы. В соответствии с Meraldi et al. (28), здесь мы демонстрируем, что статус р53 не оказывает прямого влияния на удвоение центросом в каждой из нескольких клеточных линий млекопитающих. Наоборот, подавление p53 вызывает амплификацию центросом за счет обязательного нарушения контрольной точки тетраплоидии G 1 . Важно отметить, что мы дополнительно установили, что эффект подавления р53 на амплификацию центросом неотличим от результатов, полученных при подавлении белков кармана RB.Поскольку путь белкового кармана RB, по-видимому, нарушен практически во всех опухолях (29–31), наши результаты свидетельствуют о том, что контрольная точка тетраплоидии G 1 обычно подавляется в опухолях, что делает их уязвимыми как для прогрессирования анеуплоидии, так и для амплификации центросом. На этом пути мы показываем, что кластеризация центросом в тетраплоидном митозе, который следует за уходом от тетраплоидии G 1, имеет решающее значение для процесса амплификации центросом. Наконец, наши результаты предлагают объяснение предыдущих результатов, которые, по-видимому, установили прямую связь между статусом p53 и амплификации центросом в эмбриональных фибробластах мыши (MEFs; ref.26). Мы показываем, что p53-компетентные клетки MEF, модельная система, используемая в этом исследовании, быстро прекращают цикл in vitro и становятся частично тетраплоидными. Однако, когда либо p53, либо RB-карманные белки супрессированы, MEFs проявляют такое же поведение, как мы описываем для других клеток, подвергаясь амплификации центросом в результате прохождения мимо тетраплоидных G 1 с последующим кластеризацией центросом в митозе. Материалы и методыГенерация p53−/− MEF.мыши C57B1/6J, гетерозиготные по р53, были приобретены в лаборатории Джексона. Мышей спаривали и на 13-й день беременности самку убивали. Эмбрионы промывали в PBS, удаляли голову и внутренние органы, а остаток измельчали, а затем пропускали через 1-мл шприц с иглой 18 калибра для диспергирования клеток. Клетки высевали на 10-см планшет, покрытый 0,1% желатина, и выращивали в DMEM, содержащей пен-стрептококк и 15% (об./об.) FBS (HyClone), как описано Harvey et al. (32). Клетки генотипировали с помощью ПЦР в соответствии с инструкциями из лаборатории Джексона. Культура клеток.Первичные эмбриональные фибробласты крысы (REF)-52 и их производные, трансформированные большим Т-антигеном обезьяньего вируса-40 (TAG; ссылка 33), были любезным подарком Г. Р. Старка (Кливлендская клиника, Кливленд, Огайо). Клетки IMR-90 и VA-13 были получены из репозиториев клеток Coriell (Camden, NJ). p53DD REF-52 получали путем инфицирования REF-52 мышиными ретровирусами, экспрессирующими укороченный мутант p53DD p53, как описано в Andreassen et al. (19). Тройной нокаут белка RB (TKO) MEF был любезным подарком Julien Sage и Tyler Jacks (Массачусетский технологический институт, Кембридж, Массачусетс) (34). Время удвоения клеток в середине логарифмической фазы составляло 28 ч для MEF дикого типа, 24 ч для REF-52, p53DD REF-52, IMR-90, p53 -/- MEF и TKO MEF и 20 ч для TAG. и ВА-13. Все клетки культивировали в виде монослоев в DMEM (Invitrogen) с добавлением 10% (об./об.) FCS (Biological Industries, Beit Haemek, Израиль). Клетки поддерживали во влажном инкубаторе при 37°C в среде с 5% CO 2 . Лекарственные препараты и протоколы синхронизации.Для ареста клеток на границе фаз G 1 /S клетки со случайным циклом инкубировали с 2 мМ гидроксимочевины в течение указанного времени. Чтобы вызвать нарушение цитокинеза, клетки REF-52, экспрессирующие p53DD, синхронизировали в митозе, а затем инкубировали с 10 мкМ DCB в течение 5 часов, как описано (19), а затем высвобождали в не содержащую лекарственное средство среду в течение указанного времени. Альтернативно, p53 -/- и TKO MEF подвергали воздействию 10 мкМ DCB в течение 24 часов, а затем высвобождали.Гидроксимочевина, DCB и нокодазол были получены от Sigma. Проточный цитометрический анализ.Для анализа профилей клеточного цикла клетки были подготовлены для проточной цитометрии с использованием иодида пропидия в качестве маркера содержания ДНК, как описано (35). Данные собирали с помощью проточного цитометра FACScan, а результаты анализировали с помощью программного обеспечения CELLQUEST (оба от Becton Dickinson). Для каждого образца было собрано 10 000 событий, а агрегированные клетки были исключены. Иммунофлюоресцентная микроскопия.Клетки, подготовленные для иммунофлуоресцентной микроскопии, выращивали на покровных стеклах, покрытых поли-d-лизином. Для анализа числа центросом и полюсов веретена клетки фиксировали 2% (вес/объем) параформальдегида в PBS при 37°C в течение 20 минут, а затем подвергали пермеабилизации в течение 3 минут с помощью 0,2% Triton X-100 в PBS. Клетки инкубировали с первичными антителами к γ-тубулину (Clone GTU-88, Sigma), разведенными 1/100 в PBS, содержащем 0,05% Tween-20 и 3% BSA, в течение 1 ч при 37°C во влажной камере.После трех промывок PBS клетки инкубировали в течение 30 минут с 2,5 мкг/мл FITC-конъюгированного вторичного антитела козы против мышиного IgG от The Jackson Laboratory. ДНК контрастировали добавлением 0,5 мкг/мл йодида пропидия в течение 5 мин. Клетки наблюдали с помощью микроскопа Optiphot II (Nikon), присоединенного к конфокальному аппарату лазерного сканирования MRC-600 (Bio-Rad Microscience Division, Herts, England). РезультатыОтсутствие p53 и функции семейства карманных белков RB не влияет на количество центросом.Нарушенная функция p53 была связана с амплификации центросом в клетках MEF со случайным циклом, а подавление функции p53 часто связано с анеуплоидным статусом опухолей. Аномальное количество центросом приводит к анеуплоидии за счет изменения количества полюсов веретена. Таким образом, мы сначала задались вопросом, оказывает ли нарушенная функция p53 какое-либо влияние 90–177 per se 90–178 на плоидность клеток в непрерывной культуре клеток. Функция p53 может быть подавлена либо экспрессией p53DD, доминантно-негативной формы p53, либо экспрессией большого Т-антигена SV40 в клетках REF-52, нетрансформированной линии REF. Как определено с помощью анализа FACScan (рис. 1), в содержании ДНК в клетках, экспрессирующих p53DD, и в клетках TAG после длительного культивирования не наблюдается выраженных изменений по сравнению с контрольными клетками REF-52, экспрессирующими функциональный p53. Поскольку Т-антиген подавляет функцию семейства RB-карманных белков, а также р53 (36), также очевидно, что подавление семейства RB не влияет на плоидность и, следовательно, на число центросом. Рисунок 1Отсутствие функций семейства белков p53 и RB не изменяет ни плоидность, ни число центросом в клетках фибробластов.( A ) Содержание ДНК в клетках REF-52, p53DD REF-52 и TAG со случайным циклом, проанализированное с помощью проточной цитометрии. Количество центросом ( B ) и полюсов веретена ( C ) в указанных клеточных линиях, определенное с помощью микроскопического анализа случайных циклических популяций, окрашенных γ-тубулином и PI. ( B ) Интерфазные клетки оценивали как имеющие нормальное количество центросом (1 или 2 пятна γ-тубулина; черные столбцы) или количество, превышающее два (белые столбцы). ( C ) Митотические клетки подсчитывали как имеющие либо два (черные столбцы), либо более двух полюсов веретена (белые столбцы).Как в B , так и в C значения представляют собой средние значения трех подсчетов по 100–200 клеток в каждом, ± стандартное отклонение. Очевидное сохранение эуплоидии при длительном культивировании в клетках с нарушенной функцией p53 или RB соответствует прямым микроскопическим подсчетам числа центросом во время интерфазы в различных клеточных линиях (рис. 1 B ). Небольшое количество клеток с более чем двумя различимыми центросомами, как определено иммунофлуоресценцией γ-тубулина в контрольных клетках REF-52, не усиливается отсутствием функции p53 или RB.Мы также проанализировали количество центросом в нетрансформированной линии клеток человека, IMR-90, и линии клеток, трансформированной Т-антигеном, полученной из IMR-90, VA-13 (фиг. 1 B ). В этих клетках также не наблюдается увеличения числа центросом. Увеличение числа центросом вызывает анеуплоидию, создавая более двух полюсов веретена во время митоза, вызывая случайное распределение хромосом в анафазе. Анализ количества полюсов веретена во время митоза в тех же клеточных линиях (рис. 1—) показал, что частота множественных (более двух) полюсов веретена была одинаково незначительной во всех исследованных клеточных линиях. Удвоение центросом происходит во время S фазы под контролем Cdk2 и циклинов A и E (22-25). Трансформированная клеточная линия яичника китайского хомячка, СНО, увеличивает количество центросом во время длительной остановки S фазы в присутствии гидроксимочевины (24, 25, 37), ингибитора репликации ДНК (38). Однако этот эффект не проявляется в других исследованных клетках млекопитающих. Чтобы определить, влияет ли отсутствие функции p53 на удвоение центросом, мы проанализировали количество центросом в REF-52 и в производных клетках, экспрессирующих p53DD или Т-антиген, во время длительной остановки с помощью гидроксимочевины.Результаты показывают незначительное очевидное влияние нарушенной функции p53 или RB на удвоение центросом во время длительной остановки S-фазы (Fig. 2). Рисунок 2Отсутствие функций семейства белков p53 и RB не приводит к амплификации центросом в клетках, задержанных в S-фазе. Клетки REF-52, p53DD REF-52 и TAG подвергали воздействию 2 мМ гидроксимочевины в разное время и анализировали количество центросом с использованием антител против γ-тубулина. Клетки подсчитывали как имеющие нормальное количество центросом (1 или 2 пятна γ-тубулина) или избыточное количество центросом (> 2 пятна) в каждый момент времени.Значения представляют собой средние значения трех подсчетов, по крайней мере, по 200 клеток в каждом, ± стандартное отклонение. Амплификация числа центросом происходит в клетках p53DD после неудачного расщепления.Ранее мы показали, что нарушение расщепления приводит к остановке G 1 в p53-компетентных тетраплоидных клетках, но эта остановка отменяется супрессией функции p53 в p53DD-экспрессирующих клетках (19). Подразумевается, что тетраплоидный статус будет генерировать анеуплоидию в p53-нулевых клетках из-за их неспособности арестовать в G 1 , т.к. клетки в следующем митозе будут содержать сверхштатные центросомы и множественные полюса веретена.Однако оставалось возможным, что субпопуляция клеток, избежавших тетраплоидного ареста, прошла через митоз с двумя полюсами веретена и, таким образом, скорее наследовала бы аномальные числа центросом, чем стала бы анеуплоидной. Предыдущие исследования показали, что множественные центросомы действительно могут группироваться на полюсах веретена (39, 40). Мы проверили эту возможность, обрабатывая клетки REF-52 p53DD со случайным циклом дигидроцитохалазином B (DCB), препаратом, который препятствует сборке актина и вызывает нарушение расщепления (41, 42).Через 30 ч после высвобождения из DCB почти треть популяции интерфазных клеток имела более двух центросом (рис. 3 A ), причем абсолютное число центросом со временем продолжало расти, так что к 72 га субпопуляция с четыре или более центросом были очевидны. Рисунок 3 Амплификация центросомыкоррелирует с индукцией нарушения расщепления DCB в p53-некомпетентных клетках (p53DD REF-52). В указанные моменты времени после высвобождения из DCB и после индукции нарушения расщепления центросомы метили антителами против γ-тубулина, а клетки докрашивали на ДНК с помощью PI.Интерфазные клетки ( A ) подсчитывали как имеющие 2 или менее, 3 или 4 или >4 центросом (γ-тубулиновые пятна), а митотические клетки ( B ) подсчитывали как имеющие 2 или >2 полюса веретена. Контроль относится к случайным циклическим клеткам. Гистограммы A и B представляют собой три счета по 100–200 клеток каждый; бары представляют SD. ( C ) Репрезентативные метафазные ( слева ) и среднеанафазные ( справа ) клетки через 30 ч после высвобождения из DCB, демонстрирующие биполярные веретена с кластеризацией центросом на полюсах веретена. Эти результаты свидетельствуют о том, что кластеризация центросом на одиночных полюсах действительно происходит во время митоза с образованием множественных центросом в дочерних клетках. Эта возможность была проанализирована двумя способами: ( i ) путем определения процента митотических клеток только с двумя полюсами веретена в тетраплоидном митозе, который следовал за нарушением дробления, и ( ii ) с помощью иммунофлюоресцентной демонстрации кластеризации центросом на полюсах веретена у эти клетки. Через 30 часов после высвобождения из DCB примерно 20% клеток p53DD с тетраплоидным статусом сформировали два полюсных веретена (рис.3 B ), и эта популяция приблизилась к 40% к 72 часам. Согласно иммунофлуоресцентному анализу кластеризация множественных центросом на одном полюсе веретена была обычным явлением в митотических клетках, образующих биполярные веретена (Fig. 3 C ). Присутствие множественных центросом не мешает ни образованию нормальной метафазной пластинки, ни нормальному анафазному разделению двух наборов хроматид (Fig. 3 C ). p53-/- и TKO Клетки MEF Увеличение числа центросом за счет обхода остановки естественной тетраплоидии.Вопреки доказательствам, представленным выше, было продемонстрировано, что MEF p53 -/- обычно содержат увеличенное число центросом в отсутствие экспериментальных манипуляций или введения других мутаций (26). Таким образом, мы исследовали MEF, чтобы определить, что может объяснить это исключительное поведение. p53-компетентные MEF необычны тем, что они прекращают пролиферировать примерно после восьми пассажей in vitro (34, 43, 44, и данные не показаны). Напротив, p53 -/- MEF (32) и TKO MEF с тройной делецией белков кармана RB, RB, p107 и p130 (34, 44), продолжают пролиферировать бесконечно. Анализ FACscan MEF при последовательных пассажах после введения в культуру показал, к нашему удивлению, что клетки становятся все более тетраплоидными при каждом пассаже (рис. 4 A ), так что при переходе от 5 к 8 пассажу соотношение 4N до 2N клеток увеличилось с 37 до 63%. Таким образом, по-видимому, есть две причины прекращения циклирования в MEF дикого типа. Часть популяции 2N прекращает цикл даже на ранних пассажах, о чем свидетельствует неспособность субпопуляции перейти от 2N после воздействия DCB и высвобождения нокодазола на пассаже 3 (рис.4 B ), но другая субпопуляция все чаще перестает циклироваться со статусом тетраплоидов. Рисунок 4Спонтанная индукция тетраплоидии в клетках MEF. ( A ) Содержание ДНК MEF дикого типа со случайным циклом при различных пассажах в культуре (как определено с помощью проточной цитометрии) показывает, что эти клетки становятся все более тетраплоидными между пассажами 5 и 8. Номер пассажа обозначен как «p5-p8» ( слева), а процент указывает на соотношение 4N к 2N клеток в этом пассаже (справа).( B ) Воздействие DCB на MEF дикого типа на пассаже 3 приводит к накоплению субпопуляции с 4N ДНК. Высвобождение из DCB в нокодазол не показывает дальнейшего прогрессирования до содержания ДНК> 4N, что указывает на то, что клетки MEF дикого типа имеют функционирующую контрольную точку тетраплоидии. Даже на пассаже 3 значительная субпопуляция остается 2N после воздействия DCB и нокодазола и, таким образом, не циклируется. ( C ) p53 -/- и TKO MEF также становятся все более тетраплоидными в культуре.Эти клетки пассажа 6 не останавливаются, когда становятся тетраплоидными после индукции нарушения расщепления с помощью DCB. Они продолжают делиться (данные не показаны) и переходят в гипердиплоидный статус. Самая простая интерпретация этих результатов состоит в том, что субпопуляция MEF дикого типа становится тетраплоидной по неизвестному механизму и останавливается, вероятно, из-за контрольной точки тетраплоидии. Как мы показали ранее для нескольких типов клеток (19), MEF, обработанные DCB в течение 24 часов, накапливают субпопуляцию 4N G 1 , создавая прочный блок, который сохраняется после введения нокодазола в течение 24 часов, что указывает на то, что MEF дикого типа имеют контрольную точку тетраплоидии (рис.4 В ). Напротив, p53 -/- и TKO MEF не проявляют контрольной точки тетраплоидии, но прогрессируют до гипердиплоидного статуса при высвобождении из DCB (Fig. 4 C ). Как показано выше для других клеточных линий, пассаж после ареста тетраплоидии в p53 -/- и TKO MEFs генерирует значительную субпопуляцию клеток с амплифицированным числом центросом (Fig. 5 A ). Сравнение результатов с p53 -/- и TKO MEF не выявило различий в количестве центросом, независимо от того, были ли подавлены белки p53 или RB.Статус p53, per se , не определяет напрямую число центросом в MEF, потому что в клетках TKO получается неразличимый результат. Рисунок 5Ни p53, ни статус карманного белка RB per se не вызывают амплификацию центросом в клетках MEF. ( A ) MEF дикого типа, p53 -/- и TKO MEF были проанализированы с помощью микроскопии на количество центросом на пассаже 6. ( B ) Альтернативно, p53 -/- MEF были подвергнуты воздействию 2 мМ гидроксимочевины (HU) в течение указанного времени с последующим подсчетом числа центросом.Клетки считали имеющими нормальное количество центросом: 1 или 2, 3 или 4 или >4 центросом (γ-тубулиновые пятна). Значения представляют собой среднее из трех подсчетов по 200 клеток в каждом ± стандартное отклонение. ( C ) Типичные случайные циклы TKO и p53 -/- MEF, которые сформировали биполярные веретена с кластеризацией центросом на полюсах веретена. В соответствии с этими результатами неконтролируемая амплификация центросом в MEF p53 -/- не происходит во время длительного блока S-фазы (рис.5 В ). Несмотря на то, что количество центросом увеличивается в клетках p53 -/- в отсутствие обработки гидроксимочевиной, остановка S-фазы не изменяет количество присутствующих центросом. Мы пришли к выводу, что изменение количества центросом в MEF не вызвано дерегуляцией контроля дублирования центросом во время S-фазы. Как показано выше, увеличение числа центросом, которое происходит после прохождения клеток за пределы тетраплоидии G 1 , также включает кластеризацию центросом с образованием биполярных веретен с множественными центросомами в тетраплоидных клетках, которые переходят к митозу.Как и ожидалось, множественные центросомы, сгруппированные на одном полюсе веретена в клетках с биполярными веретенами, обычны как в популяциях p53 -/- , так и в популяциях TKO (Fig. 5 C ). Примечательно, что, несмотря на дисперсную природу кластеризации центросом на полюсах веретена, такие несфокусированные полюса все еще способны образовывать функционирующие биполярные веретена. ОбсуждениеМы продемонстрировали, что подавление p53 или семейства белков кармана RB не вызывает непосредственно изменения количества центросом ни в одной из нескольких первичных клеточных линий млекопитающих.Наоборот, увеличение количества центросом происходит, главным образом, за счет неспособности арестовать контрольную точку тетраплоидии G 1 после неспособности сегрегировать геном в митозе. Тетраплоидные клетки в G 1 содержат две пары центриолей и будут содержать четыре пары при вступлении в следующий митоз. Вторым критическим элементом в генерации клеток с амплифицированным числом центросом является кластеризация множественных центросом на одном полюсе веретена. Мы демонстрируем, что кластеризация центросом обычно происходит в p53 -/- — и клетках с дефицитом белка кармана RB с множественными центросомами.В зависимости от распределения центросом в митозе клетка, которая продолжает цикл после неудачной остановки в тетраплоидном G 1 , может образовать двухполюсное, трехполюсное или четырехполюсное веретено в следующем митозе. При четырехполюсном веретене нормальное количество центросом будет восстановлено в сильно анеуплоидном потомстве. Напротив, если центросомы группируются с образованием двухполюсного веретена, тетраплоидный статус будет поддерживаться в клетке с удвоенным нормальным набором центросом. Также могут возникать все промежуточные состояния между этими сценариями, что в конечном итоге приводит к субпопуляции анеуплоидных клеток с множественными центросомами. Эквивалентная потеря G1 Тетраплоидный контроль с подавлением p53 или RB.p53 и белки кармана RB важны для контроля прогрессирования G 1 . р53 активируется в ответ на повреждение ДНК (45, 46) или после индукции тетраплоидии в G 1 (16, 17, 19) и действует выше белков кармана RB. Одной из его важных функций является трансактивация ингибитора циклин-зависимой киназы p21, что, в свою очередь, имеет решающее значение для остановки G 1 .Поскольку p21 в значительной степени подавляет фосфорилирование RB и высвобождение E2F, инактивация семейства карманных белков RB отменяет контроль p53/p21 (обзор см. ref. 29). В результате связи между p53 и контролем RB последствия уклонения от ареста тетраплоидии G 1 могут быть, в принципе, эквивалентными в клетках, супрессированных p53 или RB. Важным выводом из наших результатов является то, что подавление p53 или RB-карманных белков приводит к эквивалентному результату в отношении индукции анеуплоидии и потери центросомного контроля в клетках, которые избегают тетраплоидного контроля G 1 .Эти результаты согласуются с нашей недавней демонстрацией того, что остановка тетраплоидии в G 1 после повреждения ДНК зависит от активности p21 (47) и, таким образом, должна включать путь, связывающий функцию p53 и RB. Следовательно, мы заключаем, что контрольная точка тетраплоидии G 1 подавлена в клетках либо с нарушенной функцией р53, либо с функцией карманного белка RB. Поскольку функция RB-карманных белков подавлена в подавляющем большинстве опухолей, роль RB-карманных белков в контроле тетраплоидии создает потенциал для анеуплоидии и увеличения числа центросом практически во всех опухолевых клетках. И p53, и статус RB изменяют число центросом в результате подавления контрольной точки тетраплоидии и митотической кластеризации центросом.Мы обнаружили, что ни p53, ни подавление белка кармана RB не приводит к увеличению числа центросом во время длительной остановки S-фазы в присутствии гидроксимочевины. На основании работы в MEFs было высказано предположение, что дефицит p53 напрямую вызывает увеличение числа центросом и что это усиление, в свою очередь, вызывает анеуплоидию (26).Несколько линий доказательств опровергают эту возможность. Во-первых, мыши с нокаутом p53 являются эуплоидными и жизнеспособными (32), а во-вторых, CIN в опухолях сильно коррелирует с мутантными bub1, bubR1 (48) и APC (49), но, по-видимому, не коррелирует со статусом p53 (47). Кроме того, как мы показываем здесь, за исключением клеток MEF, клетки с дефицитом p53 остаются неотличимыми от нетрансформированных родительских клеток в отношении профиля ДНК и количества центросом во время длительного культивирования. Альтернативная возможность объяснения увеличения числа центросом в клетках с нарушенным p53 заключается в отсутствии ареста в G 1 после тетраплоидизации.Часто преодоление тетраплоидии быстро приводит к анеуплоидии (15, 19, 47). Как мы предположили (15), прогрессирование опухоли до сильно анеуплоидного состояния может происходить по крайней мере в два дискретных этапа. Во-первых, первичный контроль митоза или деления клеток не работает, что приводит к тетраплоидным дочерним клеткам. Во-вторых, недостаточность p53 или RB-зависимых механизмов надзора, которые обычно арестовывают такие тетраплоидные клетки в G 1 , приводит к аберрантным клеточным циклам. Как мы показываем здесь, прогрессирование до анеуплоидии обусловлено кластеризацией центросом на полюсах митотического веретена в тетраплоидных клетках.Когда центросомы группируются, формируется двухполюсное веретено и сохраняется тетраплоидия, но количество центросом увеличивается в клетках-потомках. Когда центросомы не могут кластеризоваться, нормальное число центросом может сохраняться в сильно анеуплоидном потомстве. Что вызывает немедленную неудачу митоза или расщепления, которая создает основу для образования анеуплоидии и изменения числа центросом в клетках с нарушением p53 или RB? Разумная возможность состоит в том, что мутации в bub1, bubR1, APC, BRCA1 или других белках, контролирующих функцию митотической контрольной точки и точность митотической сегрегации, могут создать условия для клеток с нарушенным p53 или RB, чтобы продолжить цикл после тетраплоидного статуса G 1 с очевидными последствиями для онкогенеза. Единственным исключением из требования о митотической неудаче или сбое расщепления, чтобы заложить основу для G 1 блокирования контрольной точки тетраплоидии и последующей аномалии числа центросом, являются клетки MEF, которые служили в качестве модельной системы для подтверждения утверждения о том, что р53 непосредственно контролирует число центросом ( 26). Как мы показываем здесь, MEF действительно исключительны. Они спонтанно прекращают цикл как у эуплоидных G 1 , так и у тетраплоидных G 1 примерно после восьми пассажей in vitro .Причина ареста эуплоидного G 1 остается неизвестной, как и причина нарушения митоза или расщепления, что приводит к аресту тетраплоидного G 1 . Однако результат ясен. Как анеуплоидия, так и аномальное число центросом возникают спонтанно как в p53 -/- , так и в MEF TKO, скорее всего, за счет блокирования контрольной точки тетраплоидии G 1 . Т.о., генерация аномальных количеств центросом в MEFs возникает таким же образом, как и в др. клетках млекопитающих. Последствия химиотерапии опухолей.Митотические ингибиторы, такие как винбластин, эстрамустин и таксол, которые вызывают митотический сбой и вхождение в тетраплоид G 1 , успешно используются в химиотерапии различных опухолей (50). Хотя нетрансформированные клетки останавливаются в тетраплоидных G 1 , клетки с нарушенным р53 продолжают цикл, свойство, как мы здесь показываем, разделяемое клетками с нарушенным белком кармана RB. Возможно, что способность этих препаратов убивать опухолевые клетки связана с их прогрессированием до нежизнеспособного анеуплоидного статуса.Мы показываем, что кластеризация центросом является основным явлением в ответ на увеличение числа центросом, и кластеризация может поддерживать клетки за счет многократного образования двухполюсных веретен и, таким образом, сохранения тетраплоидного статуса. Таким образом, успех митотических ингибиторов в терапии опухолей может быть связан с относительным успехом тетраплоидных клеток в кластеризации центросом, который может различаться в зависимости от типа клеток. Кластеризация привела бы к потомству клеток с множественными центросомами, но без анеуплоидии, тогда как отсутствие кластеризации привело бы к нормальным числам центросом, но к грубой анеуплоидии. БлагодарностиМы благодарим Julien Sage и Tyler Jacks за предоставление MEF TKO и Moshe Oren за предоставление клеток, продуцирующих ретровирус p53DD. Эта работа была поддержана грантами R.L.M. от La Ligue Nationale contre le Cancer (Laboratoire Labelisé) и l’Association pour la Recherche sur le Cancer (ARC). Ф.Б. была поддержана AssociationEspoir Isère contre le Cancer и Agence Nationale de Recherche sur le Sida. О.Д.Л. был поддержан le Commissariat à l’Energie Atomique и ARC. Сноски
Сокращения
Прогрессирование клеточного цикла после нарушения расщепления | Журнал клеточной биологииЦелью митоза является деление клетки на две генетически идентичные дочери.Однако ряд митотических дефектов может аннулировать существенную точность процесса. Некоторые из них, такие как естественная моноориентация хромосом, могут быть устранены действием контрольной точки, которая останавливает митотическую прогрессию до тех пор, пока все кинетохоры не будут прикреплены к веретену. Другие дефекты, такие как нарушение расщепления, трудноизлечимы. Нарушение дробления может быть вызвано дефектом актомиозинового аппарата расщепления, дефектами абсциссии средней части тела, хромосомными мостиками, меротелиально прикрепленными хромосомами, застрявшими в борозде деления, и неправильной ориентацией веретена (для обзора см. Margolis et al., 2003; Сторхова и Пеллман, 2004). Для соматических клеток млекопитающих нарушение расщепления обычно является редким событием в культуре (Piel et al., 2001) и считается редким событием in vivo . Тем не менее, если и когда происходит нарушение расщепления, считается, что оно особенно опасно для организма, потому что и набор хромосом, и число центросом удваиваются. Дополнительные центросомы сильно повышают вероятность того, что последующий митоз будет мультиполярным и хромосомы будут неравномерно распределены по множеству дочерних клеток (см. обзор Brinkley, 2001; Nigg, 2002; Sluder and Nordberg, 2004).Кроме того, дополнительный набор хромосом увеличивает шансы того, что некоторые из анеуплоидных дочерних клеток будут иметь достаточно генетической информации, чтобы быть жизнеспособными. Нарушение расщепления считается важным (если не первичным) источником амплификации центросом (Brinkley, 2001; Borel et al., 2002; Meraldi et al., 2002). Геномная нестабильность, вызванная неравным распределением хромосом, считается основной движущей силой многоступенчатого канцерогенеза (см. обзор Nigg, 2002; Sluder and Nordberg, 2004).Действительно, тетраплоидизация часто предшествует анеуплоидии в солидных опухолях (см. обзор Nigg, 2002). Учитывая предполагаемую опасность нарушения расщепления, было интересно определить, существуют ли механизмы, которые блокируют пролиферацию клеток, которые не расщепляются и становятся тетраплоидными. Представление о том, что клетки имеют р53-зависимую контрольную точку, которая блокирует размножение клеток, которые не могут делиться, возникло из-за того, что непрерывная обработка нормальных клеток цитохалазином для блокировки деления приводит к остановке клеточного цикла (Carter, 1967; Wright and Hayflick, 1972). ; Хирано и Куримура, 1974; Лохез и др., 2003). Кроме того, клетки, обработанные ингибиторами микротрубочек, в конце концов адаптируются к контрольной точке сборки веретена, выходят из митоза без деления и останавливаются в G1 (Minn et al., 1996; Lanni and Jacks, 1998). Сходные эксперименты на клетках с нарушенным путем р53 показали, что они продолжают циклирование (Minn et al., 1996; Lanni and Jacks, 1998; Andreassen et al., 2001). Наиболее ясная и явная демонстрация этой контрольной точки была получена в отчете Андреассена и др. (2001), которые использовали дигидроцитохалазин B для ингибирования цитокинеза в клетках REF52, первичной клеточной линии крысиных фибробластов.После удаления препарата они обнаружили, что тетраплоидные клетки останавливались в G1, тогда как многие мононуклеарные клетки в тех же препаратах продолжали цикл. Временно отделив остановку G1 от действия препарата, эти работники представили доказательства того, что остановка была характерна для двухъядерного состояния. Дальнейшие указания на то, что эта остановка была вызвана неудачным расщеплением, были получены из наблюдений, что экспрессия доминантно-негативного мутанта p53 позволяет двухъядерным клеткам подвергаться синтезу ДНК. Представление о том, что нормальные соматические клетки млекопитающих имеют «контрольную точку тетраплоидии», которая останавливает двуядерные клетки в G1 после нарушения дробления, было очень привлекательным для многих, в том числе и для нас самих, поскольку оно предоставляет организму логичный способ справиться с потенциально опасным и трудноизлечимым вирусом. ситуации (обзор см. в Margolis et al., 2003). Неудивительно, что этот предложенный контрольно-пропускной пункт вызвал значительный интерес и был рассмотрен как устоявшийся механизм.Однако универсальная применимость этой контрольной точки ставится под сомнение доказательствами того, что восстановление печени у живых людей и грызунов частично обусловлено пролиферацией многоядерных гепатоцитов (см. обзор Fausto and Campbell, 2003). Наш интерес к этой контрольной точке привел к тому, что мы инициировали серию экспериментов с нормальными клетками человека, иммортализованными теломеразой (hTERT-RPE1), чтобы определить, какое событие или состояние отслеживает эта контрольная точка.Использование цитохалазина D в концентрации, используемой Andreassen et al. (2001) для дигидроцитохалазина В мы по существу воспроизвели их результаты. Однако наш вывод о том, что значительный процент двуядерных синтезированных ДНК побудил нас пересмотреть связь между нарушением расщепления и остановкой G1 с использованием более низких концентраций препарата и модификации характеристик субстрата. контрольных точек клеточного цикла1. Хартвелл, Л. Х. и Вайнерт, Т.А. Контрольные точки: элементы управления, обеспечивающие порядок событий клеточного цикла. Наука 246, 629-34 (1989). 2. Рассел П. Контрольные точки на пути к митозу. Trends Biochem Sci 23, 399-402 (1998). 3. Нюберг, К. А., Майкельсон, Р. Дж., Патнэм, К. В. и Вейнерт, Т. А. К ПОДДЕРЖАНИЮ ГЕНОМА: контрольные точки повреждения ДНК и репликации. Annu Rev Genet 36, 617–56 (2002). 4. McGowan, C.H. & Russell, P. Реакция на повреждение ДНК: восприятие и передача сигналов.Curr Opin Cell Biol 16, 629-33 (2004). 5. МакГлинн П. и Ллойд Р. Г. Рекомбинационное восстановление и перезапуск поврежденных вилок репликации. Nat Rev Mol Cell Biol 3, 859-70 (2002). 6. Чжоу Б. Б. и Элледж С. Дж. Реакция на повреждение ДНК: контрольные точки в перспективе. Природа 408, 433-9 (2000). 7. Осборн А. Дж., Элледж С. Дж. и Зоу Л. Проверка на вилке: путь реакции на стресс при репликации ДНК.Trends Cell Biol 12, 509-16 (2002). 8. Бодди М. Н. и Рассел П. Контрольная точка репликации ДНК. Курс. биол. 11, Р953-Р956 (2001). 9. Бодди М. Н., Фурнари Б., Мондесерт О. и Рассел П. Контрольная точка репликации, обеспечиваемая киназами Cds1 и Chk1. Наука 280, 909-912 (1998). 10. Lindsay, H.D. et al. Специфичная для S-фазы активация киназы Cds1 определяет подпуть ответа контрольной точки у Schizosaccharomyces pombe.Гены Дев 12, 382-95 (1998). 11. Noguchi, E., Noguchi, C., Du, L.L. & Russell, P. Swi1 предотвращает коллапс вилки репликации и контролирует киназу контрольной точки Cds1. Mol Cell Biol 23, 7861-74 (2003). 12. Колоднер Р. Д., Патнэм С. Д. и Мьюнг К. Поддержание стабильности генома у Saccharomyces cerevisiae. Наука 297, 552-7 (2002). 13. Lopes, M. et al. Реакция контрольной точки репликации ДНК стабилизирует застопорившиеся вилки репликации.Природа 412, 557-61 (2001). 14. Сого, Дж. М., Лопес, М. и Фояни, М. Инверсия вилки и накопление одноцепочечной ДНК в остановленных вилках репликации из-за дефектов контрольных точек. Наука 297, 599-602 (2002). 15. Терсеро, Дж. А. и Диффли, Дж. Ф. Регуляция прогрессии вилки репликации ДНК через поврежденную ДНК с помощью контрольной точки Mec1/Rad53. Природа 412, 553-7 (2001). 16. Бартек Дж. и Лукас Дж. Киназы Chk1 и Chk2 в контроле контрольных точек и раке.Раковая ячейка 3, 421-9 (2003). 17. Noguchi, E., Noguchi, C., McDonald, W.H., Yates, J.R., 3rd & Russell, P. Swi1 и Swi3 являются компонентами комплекса защиты репликационной вилки у делящихся дрожжей. Mol Cell Biol 24, 8342-55 (2004). 18. Foss, E.J. Tof1p регулирует реакцию на повреждение ДНК во время S-фазы у Saccharomyces cerevisiae. Генетика 157, 567-77 (2001). 19. Katou, Y. et al. Белки контрольной точки S-фазы Tof1 и Mrc1 образуют стабильный комплекс, останавливающий репликацию.Природа 424, 1078-83 (2003). 20. Chan, R.C. et al. Сплоченность хромосом регулируется паралогом часового гена TIM-1. Природа 424, 1002-9 (2003). 21. Dalgaard, J. |